Clear Sky Science · pl
Warianty w MTNAP1 powodują chorobę neurodegeneracyjną przez upośledzenie stabilności mitochondriów
Dlaczego ta historia ma znaczenie dla zdrowia mózgu
Wiele rodzin przeżywa rozdzierające chwile, obserwując, jak dziecko stopniowo traci umiejętności rozwojowe bez jasnej diagnozy. To badanie ujawnia nową genetyczną przyczynę takiego stanu, prowadząc od jednego wadliwego genu do uszkodzonych „elektrowni” w komórkach mózgu, a w końcu do kurczenia się tkanki mózgowej. Zrozumienie tego łańcucha zdarzeń nie tylko daje odpowiedzi dotkniętym rodzinom, lecz także wyostrza szerszy obraz tego, jak kruche są systemy energetyczne mózgu.
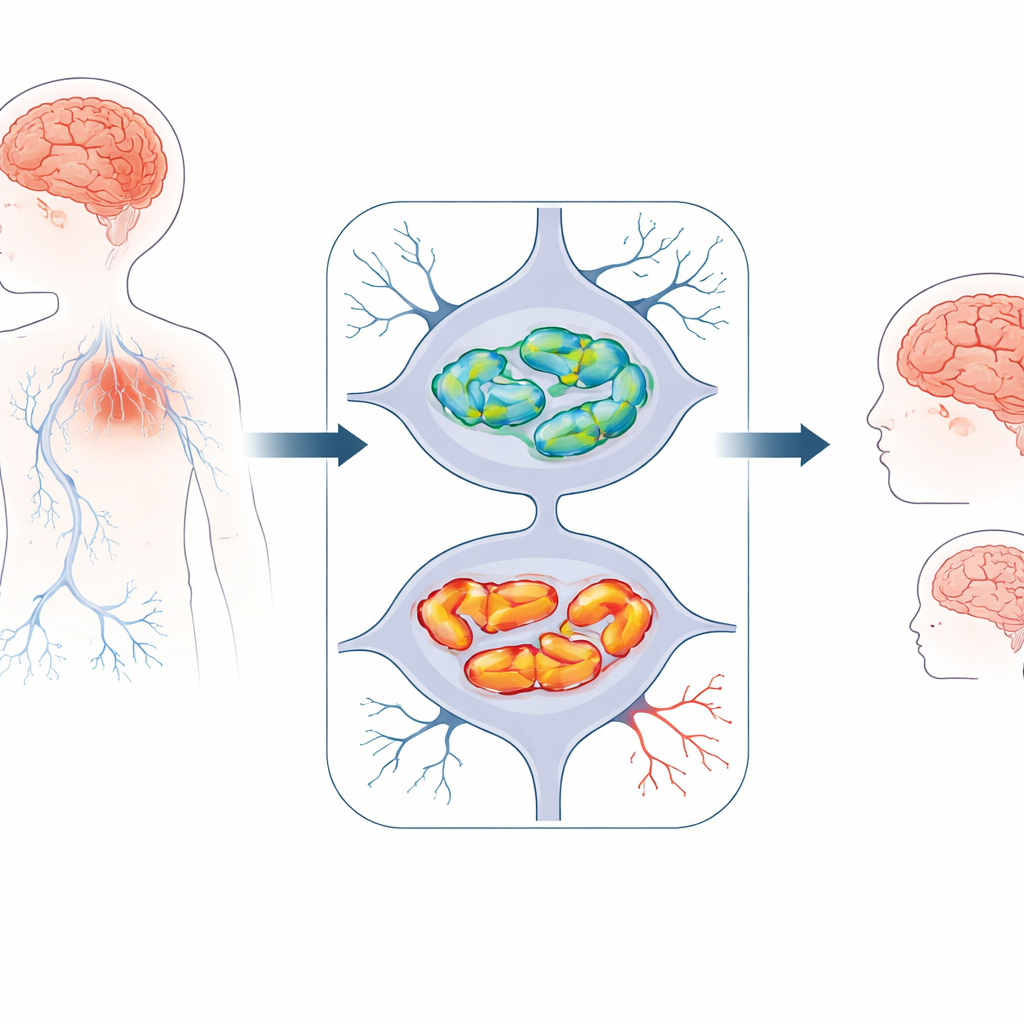
Nowo rozpoznana dziecięca choroba mózgu
Naukowcy zbadali troje dzieci z dwóch niespokrewnionych rodzin, u których występowały wczesne problemy rozwojowe. Były mniejsze niż rówieśnicy, później nabywały umiejętności siadania, chodzenia i mówienia, a następnie stopniowo traciły część tych zdolności. U wszystkich pojawiły się trudności ruchowe, takie jak chwiejny chód, sztywność mięśni lub niskie napięcie mięśniowe, oraz napady padaczkowe. Obrazowanie mózgu ukazało spójny obraz: tkanka zarówno dużego mózgu (cerebrum), jak i „małego móżdżku” z tyłu (cerebellum) cieniała z czasem, a kluczowy most łączący półkule — ciało modzelowate — był wyjątkowo wąski. Te cechy wskazywały na postępującą utratę neuronów, a nie na jednorazowe uszkodzenie przy urodzeniu.
Mały gen o wielkich konsekwencjach
Aby znaleźć przyczynę dziedziczną, zespół zsekwencjonował wszystkie geny kodujące białka u dotkniętych dzieci i ich rodziców. Skupił się na genie o nazwie MTNAP1, który pomaga organizować DNA wewnątrz mitochondriów, komórkowych fabryk energetycznych. Każde dziecko miało dwie wadliwe kopie MTNAP1, po jednej od każdego zdrowego rodzica-nosiciela. U dwojga rodzeństwa jedna „litera” w genie została zmieniona, zamieniając jeden aminokwas na inny i subtelnie zniekształcając kształt białka. U trzeciego dziecka wczesny sygnał stop prawdopodobnie uniemożliwił wytworzenie białka w ogóle. Zmiany te nie występowały w dużych bazach populacyjnych, co wzmacnia dowód, że są to rzadkie, szkodliwe warianty, a nie nieszkodliwe warianty genetyczne.
Elektrownie pod stresem
Następnie naukowcy zbadali komórki skóry pobrane od dzieci i porównali je z komórkami zdrowych osób. Pod mikroskopem normalne komórki wykazywały długie, nitkowate mitochondria tworzące połączoną sieć, podczas gdy komórki dzieci zawierały krótkie, połamane i zlepione mitochondria. Gdy badacze eksperymentalnie zmniejszyli poziomy MTNAP1 w ludzkiej linii komórek przypominających neurony, zaobserwowali ten sam rozpad sieci mitochondrialnej, potwierdzając, że utrata tego białka sama w sobie może zaburzać ich strukturę. Pomiar aktywności mitochondrialnej wykazał osłabienie kluczowych etapów produkcji energii, a komórki wytwarzały nadmiar reaktywnych form tlenu — szkodliwych produktów ubocznych działania tlenu działających jak molekularna rdza. Stresowane komórki przestały prawidłowo się dzielić, nagromadziły się w fazie spoczynkowej i włączyły markery przedwczesnego starzenia.
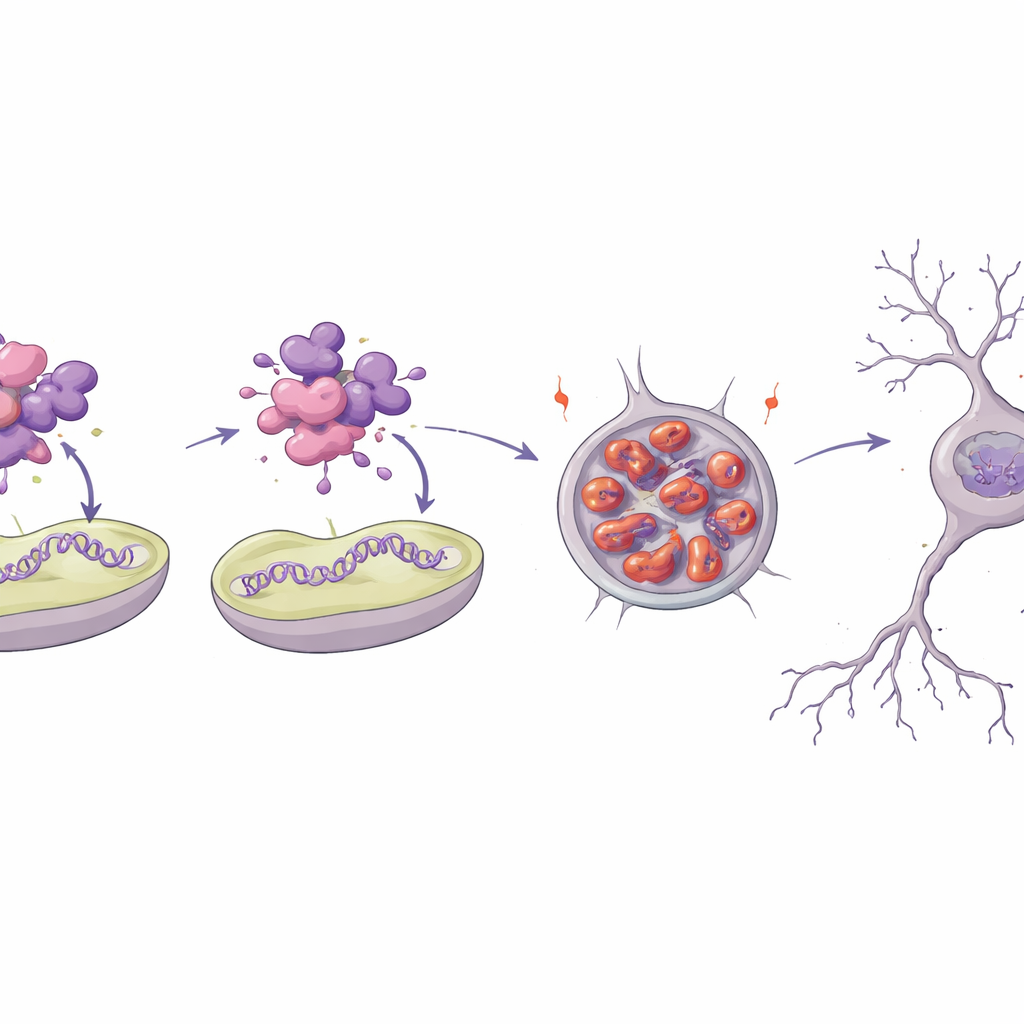
Jak jedna zmiana rozplątuje kluczowe białko
Aby zrozumieć, dlaczego jeden z wariantów jest tak niszczący, zespół modelował strukturę trójwymiarową białka MTNAP1 i odtworzył je w laboratorium. Podmieniony aminokwas znajduje się w ciasno upakowanym regionie helikalnym, który zwykle pomaga białku w interakcji z DNA mitochondrialnym i błoną wewnętrzną. Symulacje komputerowe i testy biofizyczne wykazały, że mutantyczne białko jest mniej stabilne, traci dużą część uporządkowanej struktury i łatwo tworzy grudki. W eksperymentach in vitro białko normalne silnie wiązało krótkie fragmenty DNA mitochondrialnego i sztuczne powierzchnie błonowe, podczas gdy mutant prawie wcale nie wchodził w interakcje i zamiast tego tworzył agregaty przypominające amyloid. Po wprowadzeniu do komórek przypominających neurony mutant gromadził się w dużych grudach okołojądrowych z upływem czasu, co świadczy o przeciążeniu systemów kontroli jakości białek.
Od uszkodzonych mitochondriów do zawodzącego mózgu
Składając elementy modelu, badanie przedstawia etapowy mechanizm: wadliwe MTNAP1 osłabia rusztowanie pomagające organizować DNA mitochondrialne i kotwiczyć je do błony wewnętrznej; to destabilizuje mitochondria, powodując ich fragmentację i utratę efektywności produkcji energii; narastający stres oksydacyjny i sygnały „przedwczesnego starzenia” sprawiają, że neurony stają się szczególnie wrażliwe, ponieważ mają wysokie i stałe zapotrzebowanie na energię oraz ograniczoną zdolność do odnawiania się. W rozwijającym się mózgu ten powolny, trwający kryzys energetyczny przekłada się na zahamowanie osiągania kamieni milowych, utratę nabytych umiejętności i stopniowe kurczenie się kluczowych obszarów mózgu. Choć potrzeba więcej pacjentów i badań na modelach zwierzęcych, aby w pełni odtworzyć ten zespół, praca ta mocno lokuje MTNAP1 jako kluczowego strażnika stabilności mitochondriów i podkreśla organizację DNA mitochondrialnego jako centralny filar zdrowego rozwoju mózgu.
Cytowanie: Kumar, A., Saha, S., Nasir, N. et al. Variants in MTNAP1 underlie a neurodegenerative disorder by impairing mitochondrial stability. npj Genom. Med. 11, 19 (2026). https://doi.org/10.1038/s41525-026-00554-3
Słowa kluczowe: mitochondria, neurodegeneracja, genetyka pediatryczna, DNA mitochondrialny, zlewanie białek