Clear Sky Science · pl
Wydajne i dokładne przestrzenne łączenie uczenia maszynowego potencjałów międzyatomowych dla nauki o materiałach
Dlaczego szybsze symulacje atomowe są ważne
Projektowanie lepszych materiałów do technologii takich jak fuzja jądrowa, mikroelektronika czy stopy konstrukcyjne coraz częściej opiera się na symulacjach komputerowych śledzących ruchy i oddziaływania atomów. Najdokładniejsze metody czerpią z mechaniki kwantowej, lecz są tak kosztowne obliczeniowo, że praktyczne są tylko niewielkie układy i krótkie skale czasowe. W artykule przedstawiono ML‑MIX — technikę i pakiet oprogramowania, które pozwalają zachować bliską dokładność kwantową dokładnie tam, gdzie jest ona potrzebna, używając jednocześnie prostszych, tańszych modeli w pozostałych obszarach. Efekt to istotne przyspieszenie — często rzędu 4–10 razy — bez utraty wiarygodności kluczowych przewidywań fizycznych.
Łączenie szczegółowego i uproszczonego obrazu atomów
Rdzeniem pracy jest prosta idea: nie każdy atom w symulacji wymaga takiego samego poziomu uwagi. Obszary, w których wiązania się rozciągają, zrywają lub przekształcają — na przykład defekty, powierzchnie czy wszczepione cząstki — korzystają z nowoczesnych uczonych maszynowo potencjałów międzyatomowych, które naśladują dokładność mechaniki kwantowej. Atomy daleko od tych „ognisk” głównie drgają wokół uporządkowanych pozycji i mogą być opisane znacznie prostszymi modelami. ML‑MIX zapewnia sposób łączenia dokładnego, lecz kosztownego modelu z oszczędniejszym „tanim” modelem w tej samej skrzyni symulacyjnej. Dokonuje tego poprzez zdefiniowanie strefy rdzeniowej używającej drogiego modelu, otaczającej jej strefy buforowej, gdzie siły są starannie mieszane między modelami, oraz zewnętrznej strefy masowej używającej wyłącznie taniego opisu. 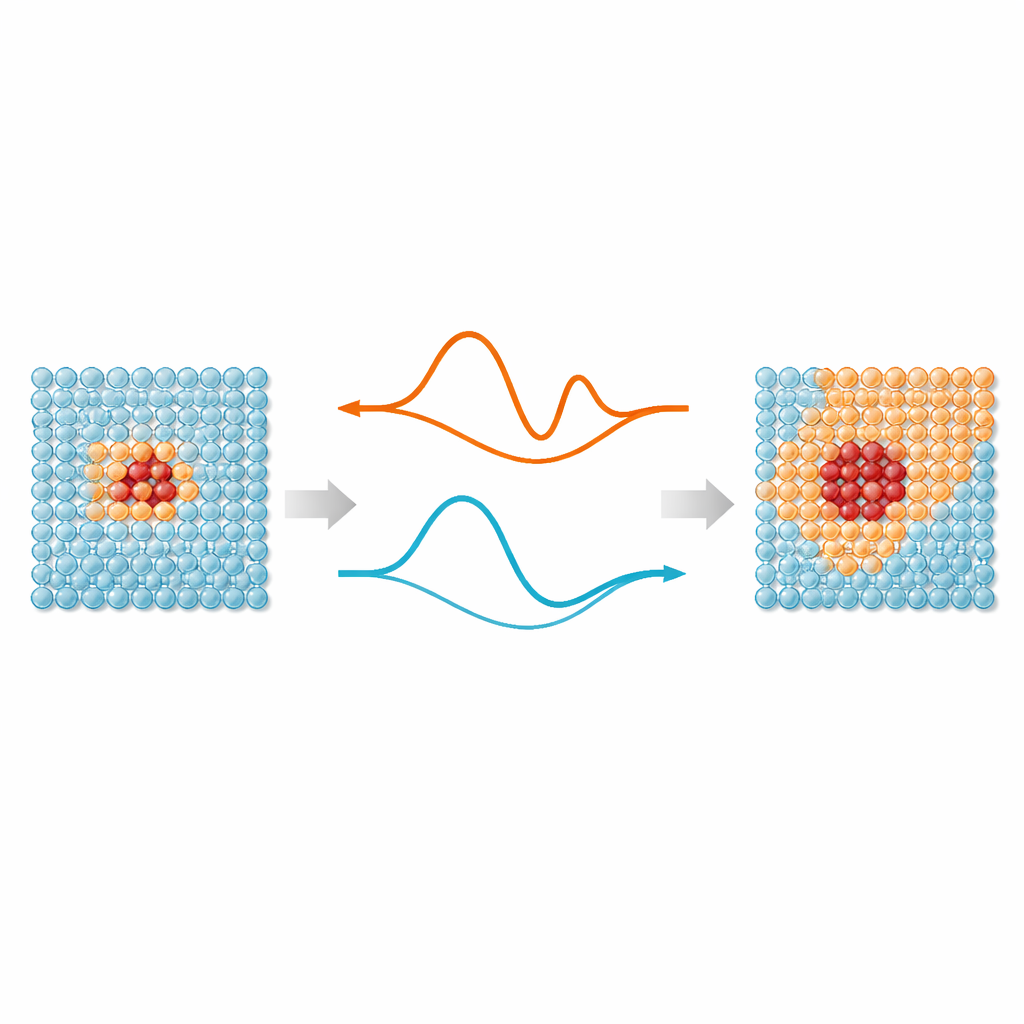
Nauczanie taniego modelu, aby naśladował dokładny
Kluczowym wyzwaniem jest zapewnienie, by tani model zachowywał się jak dokładny tam, gdzie się spotykają. Zamiast dopasowywać tani model bezpośrednio do obszernego i zróżnicowanego zestawu danych kwantowo‑mechanicznych, autorzy generują ukierunkowane „syntetyczne” dane, uruchamiając dokładny model w specyficznych warunkach istotnych dla obszaru masowego: wysokotemperaturowych drgań i łagodnie odkształconych kryształów. Następnie dopasowują tani model tak, aby odpowiadał tym danym, narzucając przy tym ścisłe ograniczenia na podstawowe własności materiału, takie jak stałe sprężystości i odstęp sieciowy. To ograniczone dopasowanie zapewnia, że naprężenia i odkształcenia długozasięgowe przechodzą płynnie przez granicę między modelami, unikając sztucznych sił, które mogłyby zafałszować dynamikę w pobliżu interfejsu.
Testowanie metody
Aby sprawdzić, czy ML‑MIX rzeczywiście działa, autorzy przeprowadzają zestaw testów dla układów krzemu, żelaza i wolframu. Dla prostego przykładu obliczają barierę energetyczną dla wakansu — pustego miejsca w sieci — w krzemie, gdy przemieszcza się z jednej pozycji na inną. Symulacja mieszana odtwarza wynik pełnej, kosztownej kalkulacji z dokładnością do tysięcznej części elektronowolta (eV), działając przy tym około pięć razy szybciej. W bardziej dynamicznym scenariuszu rozciągają pojedyncze wiązanie krzemu w gorącym krysztale i mierzą średnią siłę na nim. Symulacja używająca tylko taniego modelu już zaskakująco dobrze zbliża się do odniesienia, ale gdy dookoła rozciągniętego wiązania dodany zostanie niewielki drogi rdzeń, zgodność staje się statystycznie nieodróżnialna od w pełni dokładnego odniesienia, z przyspieszeniami sięgającymi około 13‑krotności w uruchomieniach seryjnych.
Śledzenie ruchu defektów i cząstek
Bardziej realistyczne testy badają, jak defekty poruszają się przez metale. Zespół symuluje dyfuzję defektu samowysepienia w żelazie oraz atomów helu wewnątrz wolframu. W każdym przypadku kosztowny model jest ograniczony do małego, poruszającego się obszaru wokół defektu, podczas gdy resztę kryształu opisuje tani potencjał. Uzyskane współczynniki dyfuzji zgadzają się z wynikami z pełnych, dokładnych symulacji w granicach błędu statystycznego, nawet gdy symulacja tylko z tanim modelem zawiodłaby. Autorzy następnie stosują metodę do większych, naukowo istotnych problemów w wolframie, ważnego kandydata materiału dla reaktorów fuzyjnych. Modelują ruch dyslokacji śrubowych — defektów liniowych kontrolujących odkształcenie plastyczne — oraz implantację atomów helu w gorącej powierzchni wolframu. W obu przypadkach ML‑MIX odtwarza wyniki uzyskane wyłącznie kosztownym modelem, zmniejszając koszty obliczeniowe o czynniki rzędu czterech do jedenastu. 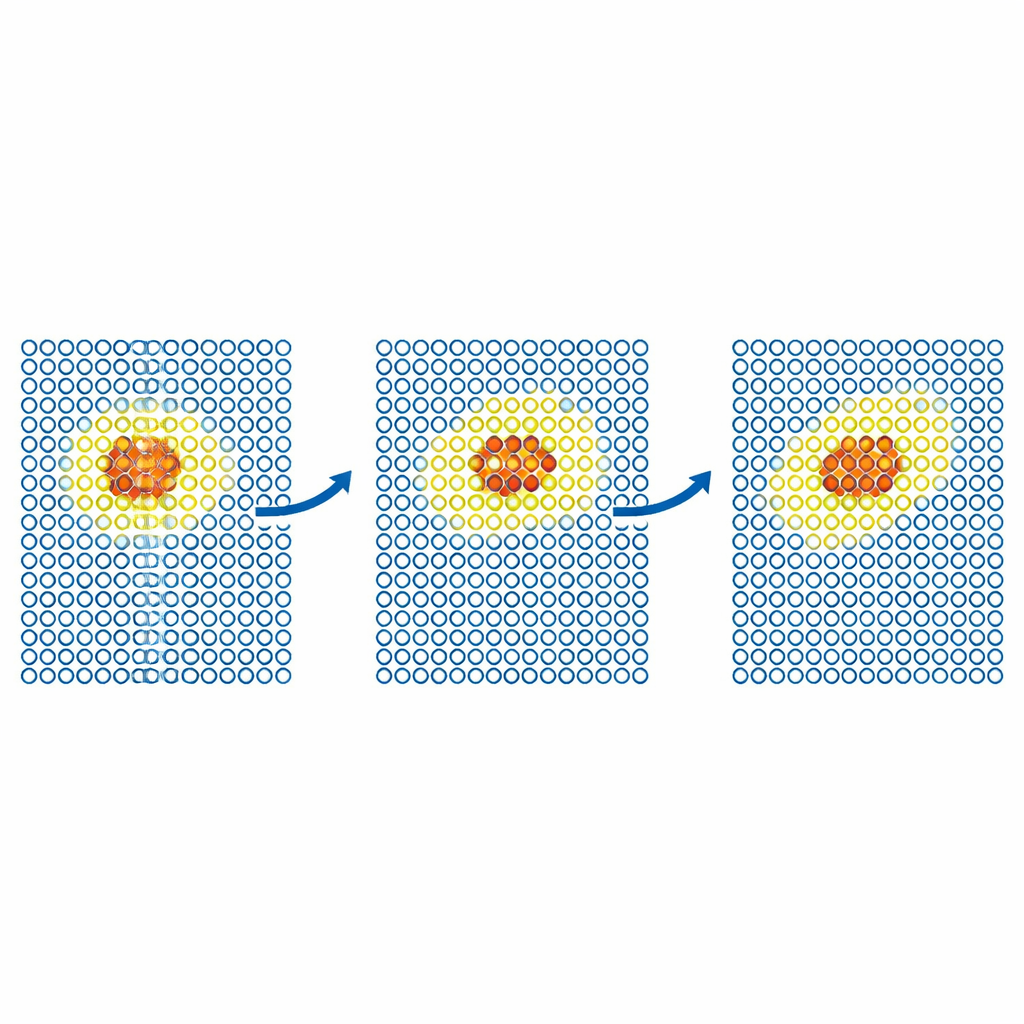
Dopasowanie do eksperymentów i perspektywy
Badanie implantacji helu najlepiej pokazuje siłę tego podejścia. Mieszając nowoczesny model uczony maszynowo dla oddziaływań hel‑wolfram z szybszym potencjałem dla czystego wolframu, autorzy symulują znacznie więcej zdarzeń uderzeń i większe próbki niż byłoby to inaczej możliwe, wszystko na procesorach graficznych. Przewidywany odsetek atomów helu, które odbijają się od powierzchni zamiast implantować wewnątrz metalu, zgadza się z pomiarami eksperymentalnymi przy energiach padających do około 80 elektronowoltów — wynik, z którym wcześniejsze symulacje miały problemy. Choć schemat miksowania nie zachowuje ściśle energii i wymaga łagodnych termostatów, powstały dryf jest niewielki i da się nim zarządzać. Ogólnie ML‑MIX pokazuje, że staranne łączenie szczegółowych i uproszczonych modeli atomowych może przełamać długo stojące bariery między dokładnością a skalą, otwierając drogę do rutynowych, wysokiej wierności symulacji złożonych materiałów w realistycznych warunkach.
Cytowanie: Birks, F., Nutter, M., Swinburne, T.D. et al. Efficient and accurate spatial mixing of machine learned interatomic potentials for materials science. npj Comput Mater 12, 110 (2026). https://doi.org/10.1038/s41524-026-01982-6
Słowa kluczowe: uczone maszynowo potencjały międzyatomowe, symulacje materiałów wieloskalowe, implantacja helu w wolframu, defekty i dyslokacje, przyspieszenie dynamiki molekularnej