Clear Sky Science · pl
Graficzne rozwinięcie klastra atomowego do podstawowych międzyatomowych potencjałów uczących się maszynowo
Nauka komputerów „czucia” atomów
Projektowanie nowych materiałów do baterii, samolotów czy reaktorów fuzyjnych często sprowadza się do prostego pytania: jak atomy na siebie oddziałują? Dokładne obliczanie tych sił jest tak kosztowne, że dla jednego materiału może trwać dni na superkomputerze. Artykuł przedstawia nową rodzinę modeli uczących się maszynowo, nazwaną GRACE, które działają jak uniwersalny „kalkulator” sił atomowych obejmujący większość układu okresowego. Celem jest uczynienie dokładnych symulacji złożonych materiałów rutyną, a nie heroicznym wysiłkiem.
Jeden model dla wielu materiałów
Większość istniejących pól sił uczących się maszynowo to narzędzia wyspecjalizowane: działają bardzo dobrze dla kilku pierwiastków lub związków, ale trzeba je budować od nowa, gdy dodaje się nowe pierwiastki. GRACE wybiera inną drogę. Został zaprojektowany od początku jako model podstawowy zdolny obsłużyć 89 pierwiastków chemicznych i ogromną różnorodność uporządkowań atomowych przy użyciu jednego, wspólnego zestawu reguł. Aby to osiągnąć, autorzy wykorzystują ramy matematyczne zwane rozwinięciem klastra atomowego i rozszerzają je do struktur przypominających grafy, co pozwala modelowi opisywać zarówno lokalne otoczenia atomów, jak i bardziej rozległe wzorce w sposób ujednolicony. Zamiast na stałe kodować każdy możliwy typ oddziaływania, GRACE uczy się zwartego „osadzenia” (embeddingów), które uchwycają podobieństwa między pierwiastkami, dzięki czemu wiedza o jednym materiale może pomóc opisać inny.
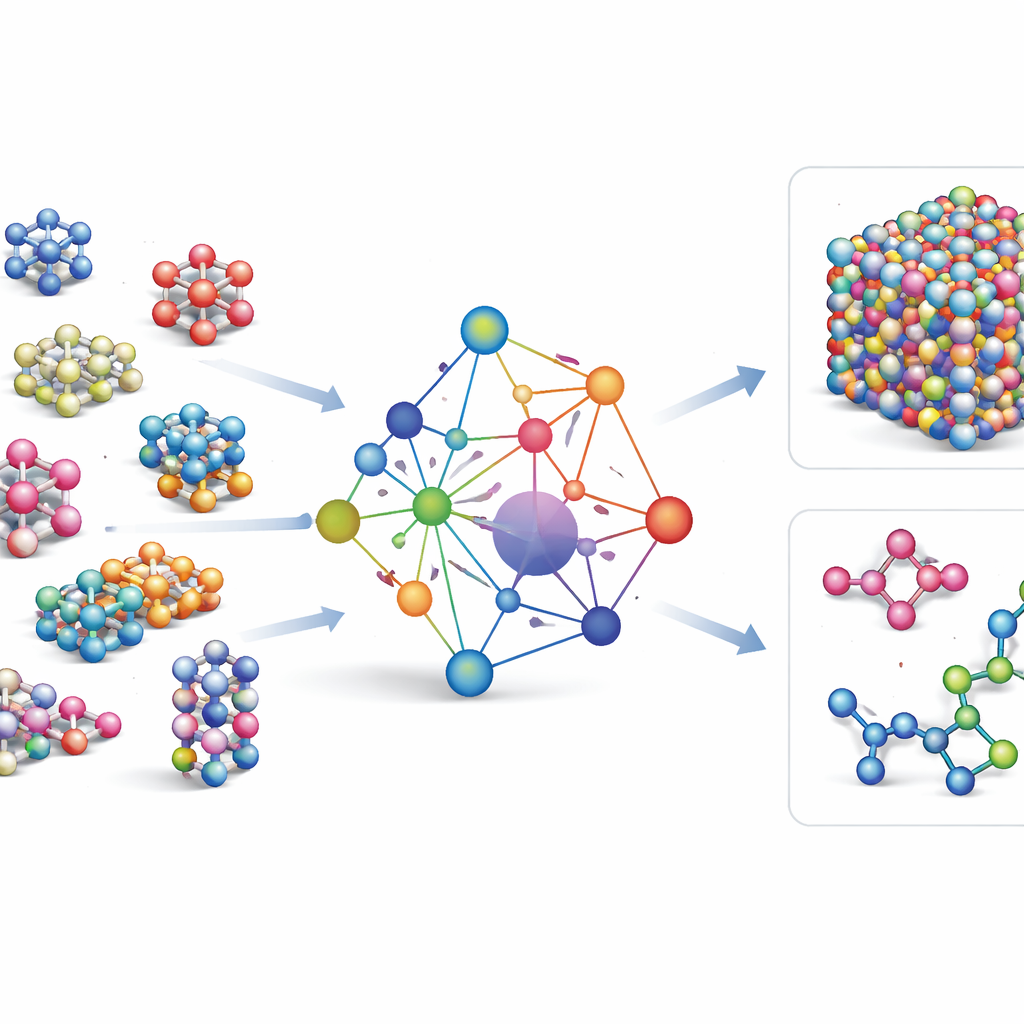
Szkolenie na morzu danych atomowych
Aby nauczyć GRACE, jak zachowują się atomy, autorzy zebrali jedne z największych publicznych baz obliczeń kwantowo-mechanicznych. Rdzeniem jest kolekcja OMat24, zawierająca około 110 milionów symulacji materiałów nieorganicznych, uzupełniona przez dwie inne bazy śledzące relaksację i ewolucję struktur. Razem te zbiory obejmują kryształy bliskie równowadze, struktury poddane odkształceniom i naprężeniom, płyny wysokotemperaturowe i inne, dla tego samego szerokiego zestawu pierwiastków. Modele GRACE występują w kilku rozmiarach, od prostszych wersji jednopoziomowych analizujących jedynie lokalne otoczenie atomowe, po głębsze wersje dwupoziomowe, które skutecznie przekazują „wiadomości” między sąsiednimi regionami. Początkowe szkolenie dąży do dobrego wyważenia energii, sił i naprężeń wewnętrznych, a dalsze dostrajanie przygotowuje modele do zgodności z powszechnie używanymi bazami odniesienia w nauce o materiałach.
Sprawdzanie modelu w praktyce
Uniwersalny model jest użyteczny tylko wtedy, gdy działa niezawodnie w wielu zadaniach. Autorzy poddali więc GRACE wymagającemu zestawowi testów odzwierciedlającemu rzeczywiste zastosowania symulacji atomistycznych. W ogólnospołecznościowym benchmarku odkrywania stabilnych struktur krystalicznych GRACE konsekwentnie znajduje się na „froncie Pareto”: dla danej dokładności jest szybszy od konkurentów, a dla danej prędkości jest dokładniejszy. Podobne korzyści pojawiają się przy przewidywaniu przewodności cieplnej, właściwości silnie zależnej od drobnych zmian w ruchu atomów. GRACE dobrze radzi sobie także z własnościami elastycznymi, energiami powierzchni, energiami granic ziaren i energiami tworzenia defektów punktowych w wielu czystych metalach — wszystkie te testy badają odpowiedź materiałów na rozciąganie, cięcie lub lokalne uszkodzenie. Długie symulacje dynamiki molekularnej gorącej roztopionej soli pokazują, że model pozostaje numerycznie stabilny przez nanosekundy, jednocześnie odtwarzając szczegółowe wzorce strukturalne i szybkości dyfuzji atomowej.
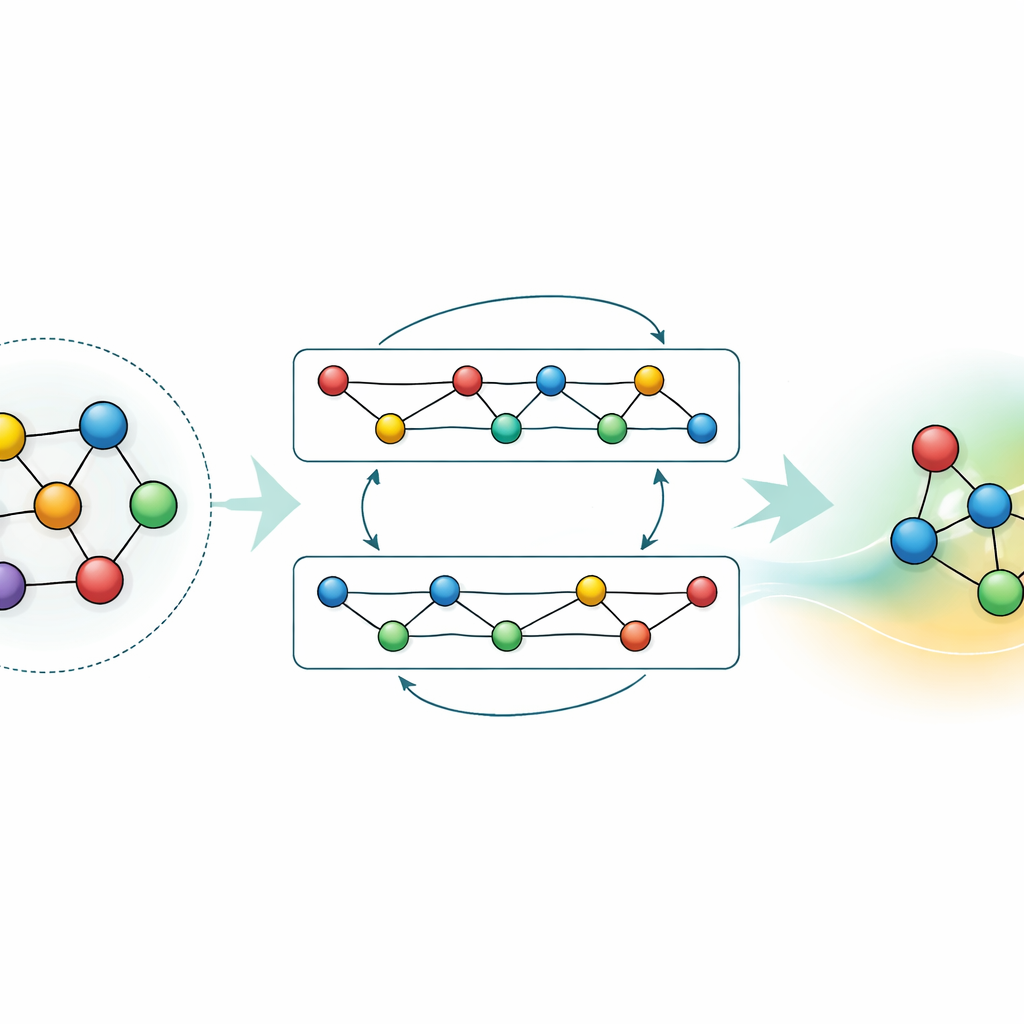
Dostosowywanie i kompresja wiedzy
Choć model ogólnego przeznaczenia jest potężny, wiele zastosowań wymaga albo większej dokładności dla konkretnego materiału, albo szybszych obliczeń na skromnym sprzęcie. Autorzy demonstrują dwie strategie osiągania tego bez utraty wiedzy zgromadzonej przez GRACE. Po pierwsze, dostrajają model podstawowy na ukierunkowanych zestawach danych, na przykład stopach aluminium–litu lub szczegółowych ścieżkach spalania wodoru. Dla stopów nawet niewielkie dodatkowe dane znacznie poprawiają przewidywania, przewyższając modele trenowane od zera na tym samym materiale. W przypadku spalania proste dostrajanie zazwyczaj powodowałoby „zapominanie” przez model informacji o innych materiałach; przez ostrożne zamrażanie części sieci i aktualizowanie jedynie wybranych parametrów autorzy ograniczają to katastrofalne zapominanie, jednocześnie uzyskując wyższą dokładność dla nowej chemii. Po drugie, pokazują, jak zdestylować duży model do znacznie prostszego „ucznia”, który na kluczowych układach naśladuje nauczyciela. Ta zdestylowana wersja działa około siedemdziesiąt razy szybciej na CPU i zachowuje większość dokładności, zwłaszcza gdy jest trenowana na mieszance złożonych stopów i prostszych struktur referencyjnych opisanych przez pierwotny GRACE.
Co to oznacza dla przyszłego projektowania materiałów
Praca stawia GRACE jako elastyczne podstawy dla następnej generacji modelowania atomistycznego. Zamiast tworzyć nowy potencjał dla każdego materiału czy właściwości, badacze mogą zacząć od uniwersalnego modelu GRACE, a następnie dostroić go lub zdestylować do swoich potrzeb, oszczędzając ogromne ilości czasu obliczeniowego i pracy ekspertów. Benchmarki pokazują, że to podejście nie tylko dorównuje istniejącym narzędziom — często je przewyższa zarówno pod względem szybkości, jak i niezawodności, zwłaszcza dla wymagających właściwości takich jak transport cieplny. Dla osób niebędących specjalistami kluczowy wniosek jest taki, że pojedynczy, dobrze zaprojektowany model uczący się maszynowo może teraz służyć jako szeroko zaufany „silnik” do wirtualnych eksperymentów obejmujących dużą część układu okresowego, przyspieszając poszukiwanie lepszych baterii, katalizatorów, stopów konstrukcyjnych i materiałów energetycznych.
Cytowanie: Lysogorskiy, Y., Bochkarev, A. & Drautz, R. Graph atomic cluster expansion for foundational machine learning interatomic potentials. npj Comput Mater 12, 114 (2026). https://doi.org/10.1038/s41524-026-01979-1
Słowa kluczowe: uczenie maszynowe międzyatomowych potencjałów, modelowanie materiałów, symulacje atomowe, modele podstawowe, graficzne rozwinięcie klastra atomowego