Clear Sky Science · pl
Samouczący się potencjał uczenia maszynowego wspomagający zautomatyzowany przepływ pracy dla wysoce wydajnego projektowania materiałów złożonych układów
Inteligentniejsze poszukiwania nowych materiałów
Projektowanie nowych materiałów przypomina poszukiwanie igły w niemal nieskończonym stogu siana. Od lepszych baterii i szybszych komputerów po bardziej wydajne lasery i potencjalne nadprzewodniki działające w temperaturze pokojowej — wiele przyszłych technologii zależy od odkrycia odpowiednich ułożeń atomów. Artykuł przedstawia sposób, który pozwala sztucznej inteligencji prowadzić większość tych poszukiwań automatycznie, dramatycznie skracając czas i koszty potrzebne do znalezienia obiecujących związków.
Dlaczego łamigłówka materiałów jest tak trudna
Właściwości ciała stałego — jak dobrze przewodzi prąd, jak jest wytrzymałe, jak reaguje na światło — zależą od rozmieszczenia atomów w trójwymiarowych wzorcach zwanych strukturami krystalicznymi. W teorii można użyć mechaniki kwantowej, by obliczyć, które ułożenia są stabilne i jakie będą ich właściwości. W praktyce obliczenia kwantowe są tak kosztowne obliczeniowo, że tylko niewielką część wszystkich możliwych materiałów można sprawdzić. Trudność rośnie gwałtownie, gdy w grę wchodzi więcej niż dwa pierwiastki chemiczne, ponieważ liczba kombinacji i możliwych ułożeń atomów eksploduje, czyniąc poszukiwanie na ślepo niemożliwym.
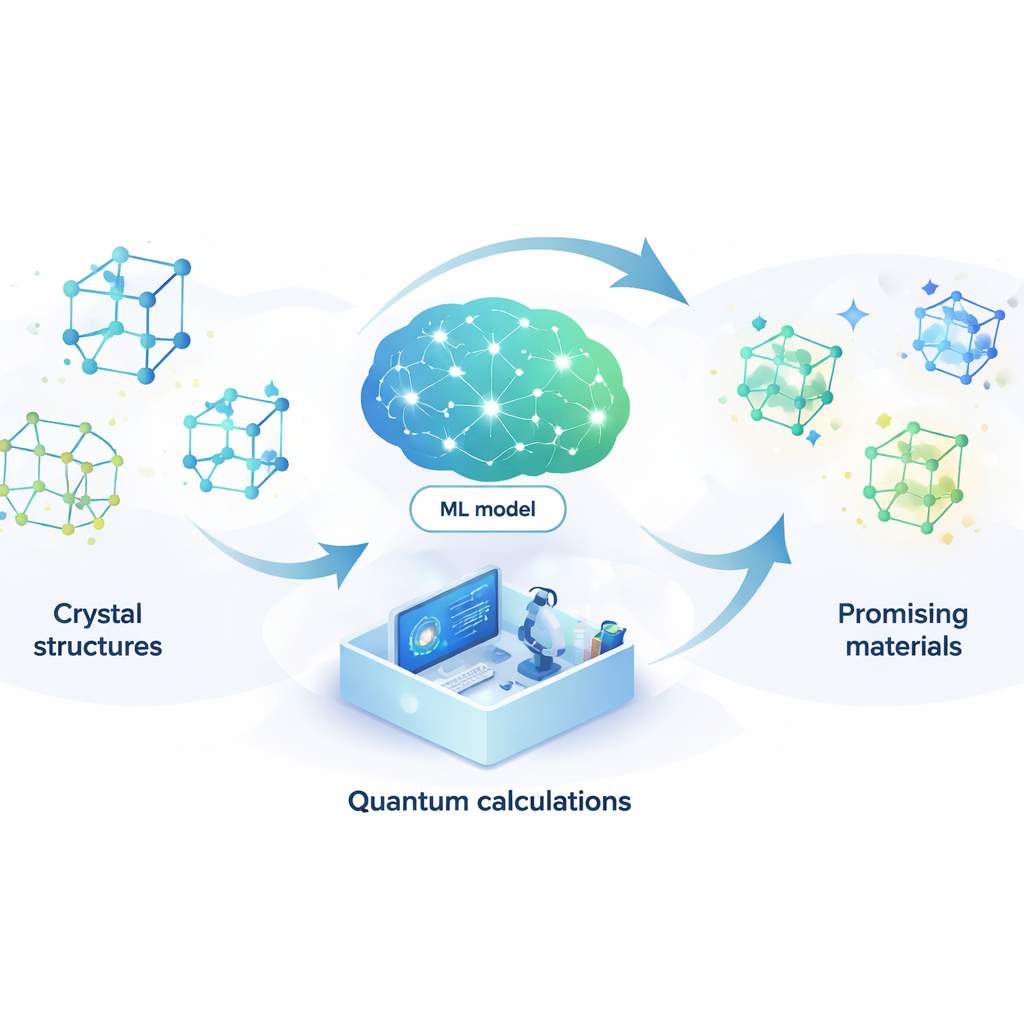
Poleganie na modelu uczącym się zamiast na fizyce kwantowej
Aby sprostać temu problemowi, autorzy zbudowali model uczenia maszynowego, który potrafi naśladować wyniki kosztownych obliczeń kwantowych przy ułamku ich kosztu. Model, zwany siecią neuronową sprzężoną z mechanizmem uwagi (ACNN), uczy się, jak energia materiału zależy od położeń i rodzajów atomów. Po przeszkoleniu potrafi bardzo szybko oszacować, czy zaproponowana struktura krystaliczna prawdopodobnie będzie stabilna oraz jakie siły działają na poszczególne atomy. Co kluczowe, model zaprojektowano tak, aby respektował podstawowe wymagania fizyczne, takie jak fakt, że przesunięcie lub obrót całego kryształu nie powinny zmieniać jego energii całkowitej.
Samo-udoskonalająca się pętla odkrywania materiałów
Zamiast trenować model jednorazowo i liczyć, że sprawdzi się wszędzie, autorzy opakowali go w pętlę samoptymalizującą się. Proces zaczyna się od niewielkiego zestawu losowych struktur krystalicznych, które są oceniane pełnymi obliczeniami mechaniki kwantowej i używane do wytrenowania początkowego ACNN. Ten model następnie relaksuje miliony próbnych struktur, szybko znajdując lokalne minima energii — kandydatów na stabilne lub prawie stabilne fazy. Przepływ pracy automatycznie wyróżnia dwa szczególnie wartościowe rodzaje struktur: te, które wyglądają na bardzo stabilne, oraz te, które wydają się niefizyczne lub podejrzane. Tylko te wybrane przypadki są ponownie wysyłane do kosztownego solvera kwantowego, a nowe wyniki są włączane do zbioru treningowego modelu. Z rundy na rundę model staje się coraz dokładniejszy w tych obszarach przestrzeni struktur, które mają największe znaczenie.
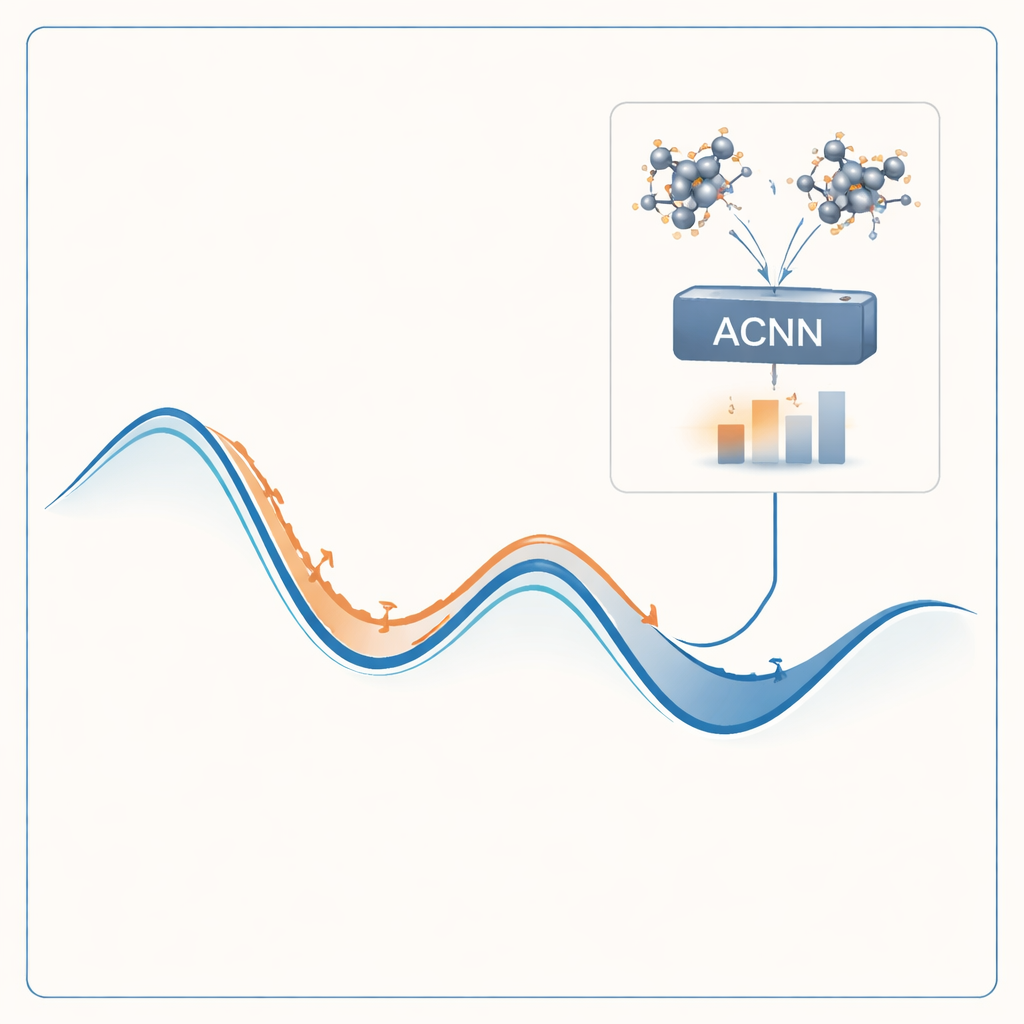
Próba metody w praktyce
Zespół zilustrował swoje podejście na dwóch wymagających układach. Pierwszy to mieszanka pod wysokim ciśnieniem magnezu, wapnia i wodoru — rodzina związków budzących duże zainteresowanie ze względu na nadprzewodnictwo w wysokich temperaturach. Eksplorując niemal sześć milionów próbnych struktur, ich przepływ pracy odkrył nową stabilną fazę MgCa₃H₂₃ oraz kilka pokrewnych struktur „klatkowych” bogatych w wodór. Obliczenia sugerują, że niektóre z tych faz mogłyby wykazywać nadprzewodnictwo przy temperaturach powyżej punktu wrzenia ciekłego azotu pod ekstremalnym ciśnieniem. Drugi test dotyczył układu czteroelementowego zawierającego beryl, fosfor, azot i tlen, wybranego ze względu na potencjał tworzenia kryształów efektywnie konwertujących światło lasera na głęboki ultrafiolet. W tym przypadku metoda zrelaksowała ponad dziewięć milionów struktur i zidentyfikowała trzy fazy termodynamicznie stabilne o bardzo szerokich przerwach energetycznych i obiecujących właściwościach optycznych.
Od brutalnej siły do ukierunkowanego odkrywania
W obu przykładach zautomatyzowany przepływ pracy osiągnął przyspieszenia rzędu około dziesięciu tysięcy razy w porównaniu z użyciem samych obliczeń kwantowych, przy jednoczesnym wiarygodnym wskazywaniu struktur wartych bliższego zbadania. Dla nietechnicznego czytelnika najważniejszy wniosek jest taki, że dużą część odkrywania materiałów można dziś powierzyć systemowi uczącemu się, który sam wykrywa obszary niepewności i prosi o ukierunkowane, wysokoprecyzyjne obliczenia tylko wtedy, gdy jest to konieczne. Tego rodzaju samokorygujące się wyszukiwanie wspomagane przez AI otwiera drzwi do badania znacznie bardziej złożonych mieszanek pierwiastków niż wcześniej, zwiększając szanse na odkrycie nowych nadprzewodników, kryształów optycznych i innych funkcjonalnych materiałów leżących u podstaw technologii następnej generacji.
Cytowanie: Li, J., Feng, J., Luo, J. et al. Self-optimizing machine learning potential assisted automated workflow for highly efficient complex systems material design. npj Comput Mater 12, 101 (2026). https://doi.org/10.1038/s41524-026-01971-9
Słowa kluczowe: odkrywanie materiałów, potencjały uczenia maszynowego, predykcja struktury krystalicznej, nadprzewodzące wodorowce, nieliniowe kryształy optyczne