Clear Sky Science · pl
Nauka efektywnej danych uproszczonej dynamiki molekularnej z sił i szumu
Dlaczego zmniejszanie modeli molekularnych ma znaczenie
Symulowanie niespokojnego ruchu każdego atomu w białku i otaczającej je wodzie jest jednym z najlepszych narzędzi do zrozumienia, jak działa życie na skali molekularnej. Jednak takie symulacje atomowe są tak wymagające obliczeniowo, że śledzenie białka podczas fałdowania, rozfałdowywania czy interakcji z partnerami przez biologicznie istotne czasy może trwać miesiącami na superkomputerze. Artykuł przedstawia nowy sposób budowy szybkich, uproszczonych modeli białek, które nadal zachowują się podobnie do pełnych modeli atomowych, przy znacznie mniejszym zapotrzebowaniu na dane treningowe i moc obliczeniową.
Od każdego atomu do prostszego obrazu
Tradycyjna dynamika molekularna śledzi każdy atom i oblicza siły między nimi w każdym drobnym kroku czasowym. Aby przyspieszyć obliczenia, naukowcy często stosują modele uproszczone (coarse-grained), które grupują wiele atomów w mniejszą liczbę „paczek” (beads). Te zredukowane modele działają znacznie szybciej, ale historycznie miały trudności z osiągnięciem dokładności symulacji atomowych, zwłaszcza dla białek o złożonym zachowaniu fałdowania. Ostatnie prace zwróciły się ku uczeniu maszynowemu, by automatycznie odkrywać lepsze pola sił w modelach uproszczonych, lecz trenowanie takich modeli zwykle wymagało milionów szczegółowych migawków, z każdą oznaczoną siłami na poszczególnych atomach — co stanowi ogromne obciążenie danych i obliczeń.
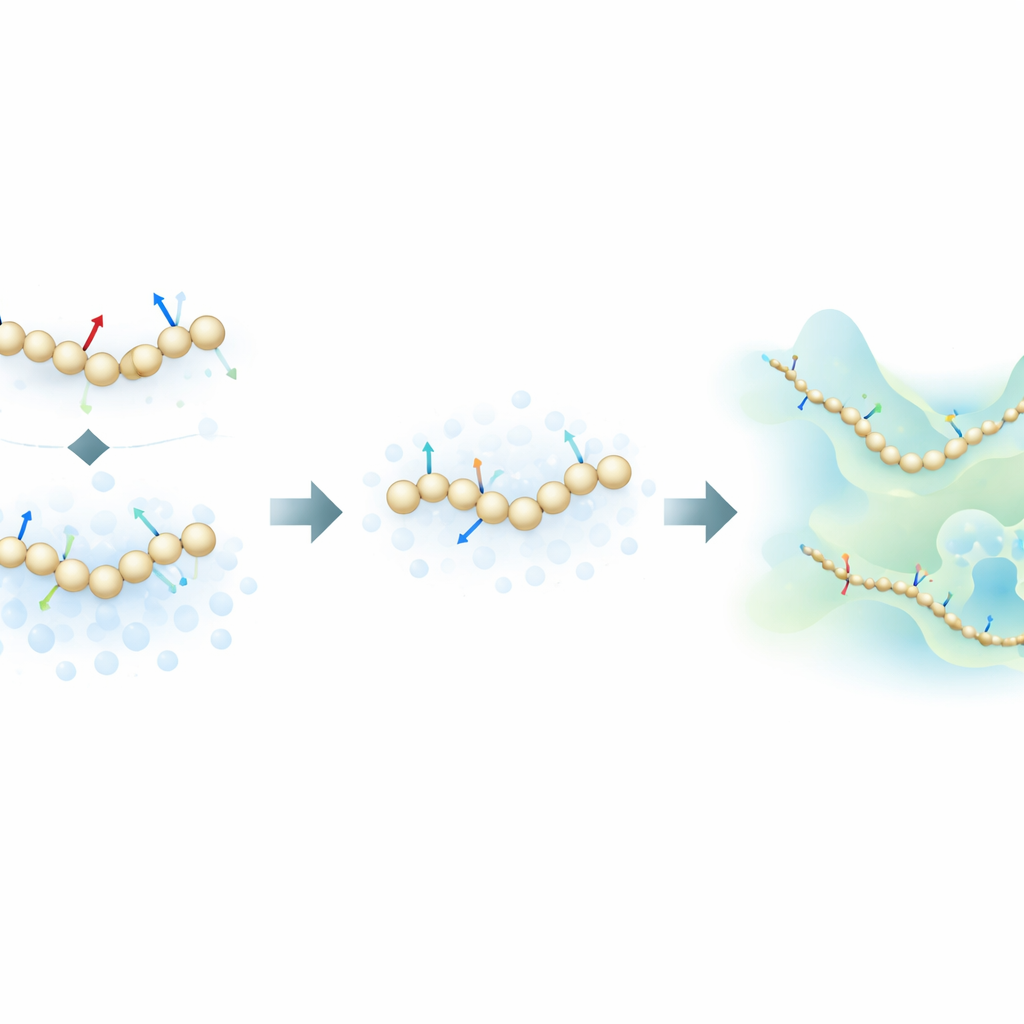
Mieszanie sił fizycznych z informacyjnym szumem
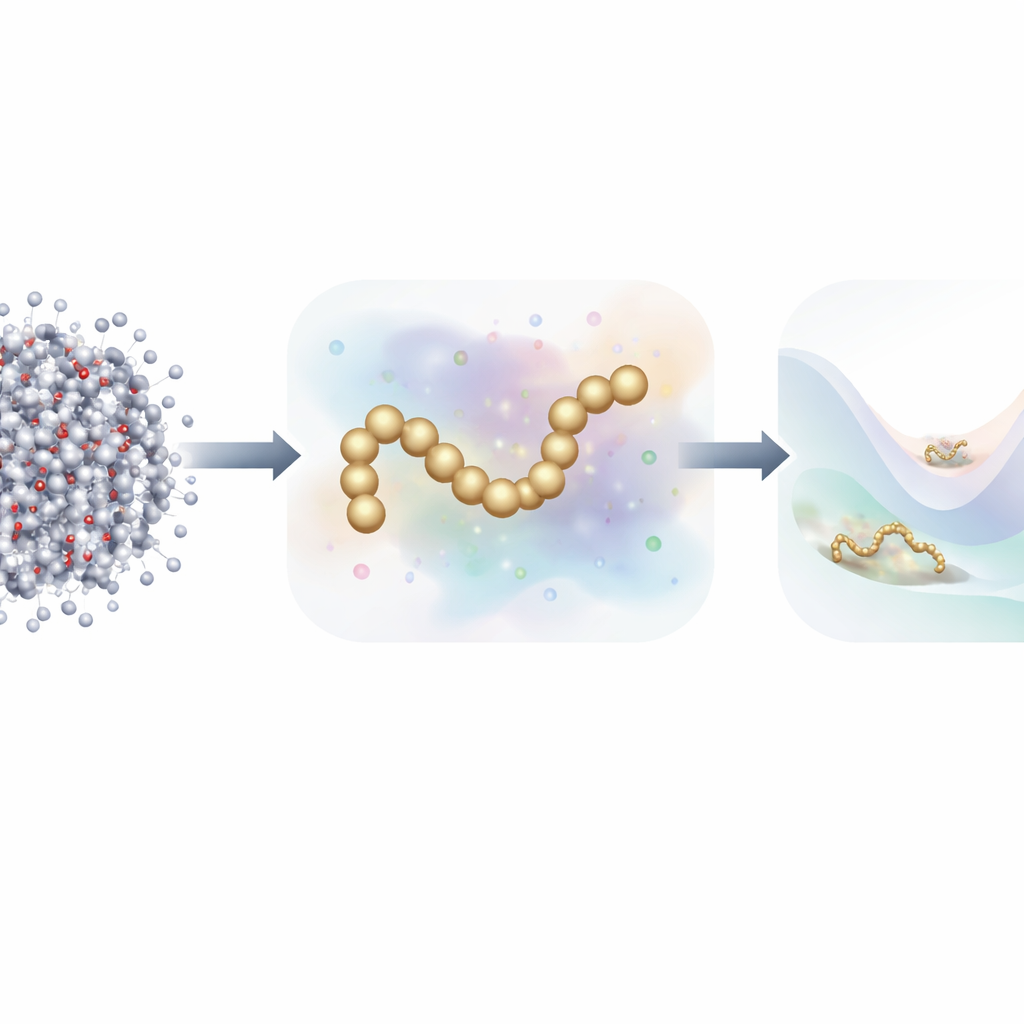
Autorzy proponują nową strategię treningową inspirowaną generatywnymi modelami dyfuzyjnymi — tą samą klasą algorytmów, która stoi za wieloma współczesnymi generatorami obrazów AI. Zamiast uczyć się wyłącznie ze skończonych sił obliczanych w symulacjach atomowych, ich metoda uczy się także z rozkładu konfiguracji molekularnych w przestrzeni poprzez celowe dodawanie kontrolowanego szumu do konfiguracji uproszczonych. W tym ujęciu szum nie jest jedynie przeszkodą do usunięcia; staje się dodatkowym źródłem informacji. Poprzez matematyczne zunifikowanie tradycyjnego podejścia „dopasowania sił” (force matching) z technikami odszumiania z modeli dyfuzyjnych, metoda potrafi wydedukować leżący u podstaw krajobraz energetyczny białka, używając znacznie mniejszej liczby oznaczonych przykładów.
Nauczanie prostych modeli naśladowania złożonych białek
Aby przetestować pomysł, badacze trenowali sieci neuronowe dla modeli uproszczonych kilku białek o rosnącej złożoności: małych minibiałek Chignolin i Trp-Cage, nieco większego NTL9 oraz 76-aminokwasowego Ubiquitinu. Porównali trzy tryby treningu: użycie tylko sił atomowych, użycie tylko informacji opartych na szumie oraz połączenie obu. Dla mniejszych białek pokazali, że nowe podejście łączone potrafi odtworzyć kluczowe cechy krajobrazu fałdowania — takie jak względna stabilność stanów złożonych i niezłożonych oraz obecność stanów pośrednich — używając do stu razy mniej danych treningowych niż standardowe metody dopasowania sił. Co zaskakujące, w reżimach ubogich w dane modele trenowane tylko na informacji pochodzącej ze szumu często dorównywały lub przewyższały dokładność treningu opartego wyłącznie na siłach.
Sięgając po większe i trudniejsze układy białkowe
Ubiquitin stanowi bardziej wymagający test: uchwycenie jego fałdowania i rozfałdowywania w realistycznych temperaturach historycznie wymagało wyspecjalizowanego sprzętu i wyjątkowo długich symulacji atomowych. Tutaj autorzy trenują modele uproszczone używając skromnego zestawu danych składającego się z krótkich symulacji w stanie równowagi wokół stanu złożonego oraz nieustalonych symulacji „ciągniętych”, które na siłę rozciągają białko. Pomimo tego stronniczego zbioru treningowego i braku idealnego odniesienia atomowego w tych samych warunkach, model trenowany z użyciem zarówno sił, jak i szumu odtwarza realistyczny obraz, w którym stany złożone i niezłożone współistnieją, przy czym stan złożony jest stabilniejszy. Dla kontrastu, model trenowany tylko na siłach nie utrzymuje stanu złożonego wcale, podczas gdy model oparty wyłącznie na szumie preferuje struktury rozciągnięte. Co warte odnotowania, żaden z modeli uproszczonych nie pamięta po prostu ekstremalnych, rozciągniętych kształtów z danych treningowych, co wskazuje, że wyuczony krajobraz energetyczny ma sens fizyczny, a nie jest jedynie odciskiem trajektorii wejściowych.
Co to oznacza dla przyszłych symulacji
Przekształcając szum w sygnał treningowy i łącząc go z siłami fizycznymi, ta praca pokazuje, że dokładne modele uproszczone białek można budować z dużo mniejszych i mniej idealnych zestawów danych niż sądzono wcześniej. W praktyce oznacza to, że badacze mogą nie potrzebować już milisekundowych symulacji atomowych na wyspecjalizowanych superkomputerach, zanim będą mogli badać zachowanie biomolekuły za pomocą modeli uproszczonych uczonych maszynowo. Zamiast tego wystarczą skromniejsze symulacje na szeroko dostępnych platformach sprzętowych, aby wytrenować potężne zredukowane modele, które uchwycą kluczowe ścieżki fałdowania i równowagi termodynamiczne. Choć pozostają pytania o to, jak najlepiej dobierać i interpretować dodawany szum oraz jak metoda sprawdzi się na jeszcze większych, bardziej złożonych zespołach biomolekularnych, podejście to znacząco obniża próg wykorzystania symulacji uproszczonych opartych na danych jako rutynowego narzędzia w naukach molekularnych.
Cytowanie: Durumeric, A.E.P., Chen, Y., Pasos-Trejo, A.S. et al. Learning data-efficient coarse-grained molecular dynamics from forces and noise. Nat Commun 17, 2493 (2026). https://doi.org/10.1038/s41467-026-70818-0
Słowa kluczowe: uproszczona dynamika molekularna, pola sił oparte na uczeniu maszynowym, symulacje fałdowania białek, modele dyfuzji w chemii, symulacje oszczędzające dane