Clear Sky Science · pl
Metrologiczna podstawa dla absolutnej transkryptomiki z zastosowaniem kalibratorów powiązanych z jednostkami Układu SI
Dlaczego przekształcanie sygnałów RNA w prawdziwe liczby ma znaczenie
Nowoczesne testy genetyczne potrafią odczytać, które geny są w komórkach aktywne, a które nie, ale mają problem z podstawowym pytaniem: ile cząsteczek faktycznie tam jest? Obecne technologie sekwencjonowania RNA w większości porównują zmiany względne między próbkami, zamiast dostarczać twardych, wiarygodnych zliczeń. To problem, jeśli chcemy ustalać uniwersalne progi chorobowe, porównywać wyniki między szpitalami lub budować precyzyjne modele działania komórek. Badanie to przedstawia nowy sposób powiązania sekwencjonowania RNA z tymi samymi międzynarodowymi jednostkami stosowanymi w chemii i fizyce, przekształcając nieostre sygnały względne w absolutne, porównywalne liczby.
Trudności w porównywaniu aktywności genów
Sekwencjonowanie RNA działa poprzez rozbijanie cząsteczek RNA na fragmenty i zliczanie, ile razy reprezentowany jest każdy gen. Jednak pojawiają się dwa rodzaje zniekształceń. Po pierwsze, systemowe różnice między eksperymentami — takie jak różne laboratoria, urządzenia czy metody przygotowania próbek — tworzą „efekty partii” sprawiające, że ta sama próbka wygląda inaczej przy powtórzeniu pomiaru. Po drugie, efekty zależne od sekwencji — gdzie geny o określonej długości lub składzie zasad są bardziej lub mniej prawdopodobne do wychwycenia — oznaczają, że nawet w obrębie jednej próbki niektóre geny są systematycznie przeszacowane, a inne niedoszacowane. W rezultacie naukowcy są w dużej mierze zmuszeni mówić o zmianach względnych między warunkami, a same te zmiany mogą wprowadzać w błąd między różnymi partiami.
Nowy zestaw miar dla pomiarów RNA
Aby to naprawić, autorzy stworzyli TranScale, panel 100 syntetycznych cząsteczek RNA zaprojektowanych tak, by zachowywać się jak prawdziwe ludzkie transkrypty, pozostając jednocześnie komputerowo odrębnymi. Te standardy obejmują szeroki zakres długości, cech sekwencji oraz klinicznie istotnych wariantów, takich jak formy splice czy fuzje genów, wiernie odzwierciedlając różnorodność rzeczywistego RNA komórkowego. Co kluczowe, każdej cząsteczce TranScale przypisano dokładne stężenie mierzone techniką pierwotną zwaną spektrometrią mas z rozcieńczeniem izotopowym, śledzalną do Międzynarodowego Układu Jednostek (SI). Poprzez dodanie znanej, bardzo małej ilości TranScale do każdej próbki RNA przed sekwencjonowaniem, eksperyment zyskuje wewnętrzną miarę, która przechodzi przez te same etapy laboratoryjne i doznaje tych samych zniekształceń co naturalne RNA.
Przekształcanie zaszumionych odczytów w absolutne zliczenia
Gdy TranScale jest obecny w każdej bibliotece, zespół może porównać liczbę odczytów sekwencjonowania dla każdej cząsteczki spike-in z jej certyfikowanym stężeniem. Dla każdej partii wybierają dobrze zachowujące się spike-iny i dopasowują liniową krzywą kalibracyjną łączącą jednostki oparte na odczytach z rzeczywistą liczbą cząsteczek. Ten prosty model jednocześnie uchwyca zarówno ogólne zniekształcenia związane z partią, jak i zależne od sekwencji. Ta sama krzywa jest następnie stosowana do wszystkich genów w próbce, konwertując ich względne odczyty na absolutne liczby kopii na jednostkę RNA. W dużym, wielo-laboratoryjnym i multi-platformowym badaniu, zaprojektowanym tak, by wywołać silne efekty partii, ta kalibracja zmniejszyła medianę zmienności pomiarów absolutnych między ośrodkami z ponad 85% do poniżej 15–25% i przywróciła poprawne grupowanie próbek biologicznych, które było wcześniej zniekształcone przez szum techniczny.
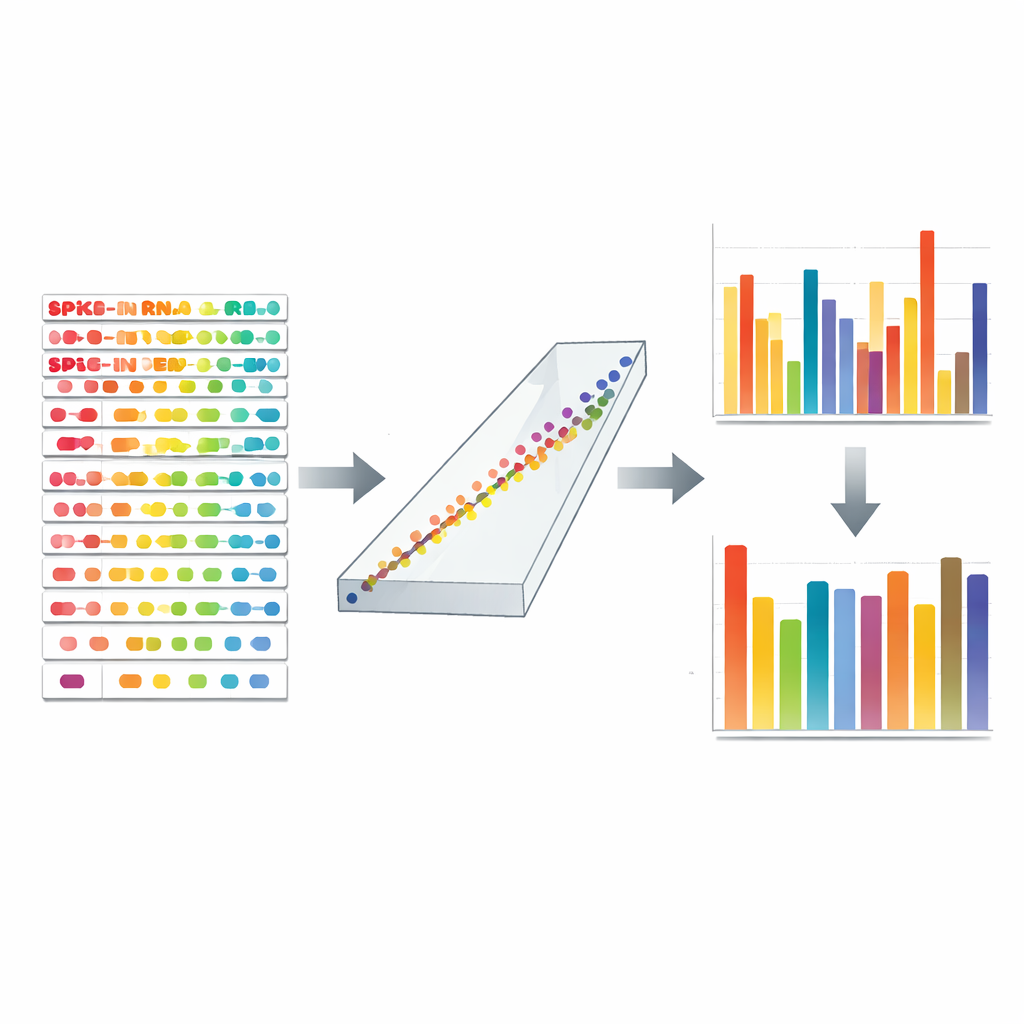
Wykrywanie ukrytych błędów i ich naprawa
Standardy TranScale pełnią też rolę diagnostycznych sond jakości danych. Poprzez porównanie zmierzonych wartości z ich certyfikowanymi wartościami, autorzy rozdzielili dwa rodzaje błędów: jak bardzo błędny jest absolutny poziom każdego genu oraz jak bardzo błędne są stosunki między warunkami. Znaleźli zaskakujące przykłady, gdzie różnice względne wydawały się spójne, ale liczby absolutne były mocno zniekształcone, i odwrotnie. Oznacza to, że konwencjonalne kontrole skupiające się wyłącznie na zmianach względnych mogą przeoczyć poważne problemy. Po kalibracji zarówno poziomy absolutne, jak i stosunki spike-inów oraz tysięcy prawdziwych ludzkich genów zgadzały się ściśle z niezależnymi pomiarami cyfrowego PCR i zewnętrznym zbiorem odniesienia. Skorygowane dane ujawniły znacznie czytelniejszy obraz ilościowy, umożliwiając porównanie genów housekeeping z genami napędzającymi raka na tej samej skali absolutnej oraz powiązanie zmian w DNA, takich jak współwzmacniane geny nowotworowe, bezpośrednio z ich produktami RNA.
Od trendów względnych do progów klinicznych
Na koniec badacze pokazali, jak skalowanie absolutne może usprawnić decyzje medyczne. Używając onkogenu często mierzonego w raku piersi, zdefiniowali stały próg oparty na cyfrowym PCR i sprawdzili, czy sekwencjonowanie RNA może niezawodnie klasyfikować próbki jako normalne lub nowotworowe w wielu partiach. Dane niekorygowane dawały niejednoznaczne odpowiedzi z powodu efektów partii. Po kalibracji TranScale każda biblioteka zgadzała się z prawdziwą klasyfikacją. Poprzez powiązanie sekwencjonowania RNA z jednostkami SI za pomocą standardów biomimetycznych, praca ta tworzy metrologiczną podstawę dla transkryptomiki. Otwiera drogę do uniwersalnych progów diagnostycznych, solidnej wymiany danych między ośrodkami oraz bardziej precyzyjnych, systemowych modeli ekspresji genów w zdrowiu i chorobie.
Cytowanie: Zhang, Y., Yang, B., Yu, Y. et al. A metrological foundation for absolute transcriptomics using International System of Units-anchored calibrators. Nat Commun 17, 2747 (2026). https://doi.org/10.1038/s41467-026-70582-1
Słowa kluczowe: sekwencjonowanie RNA, absolutna ilościowa ocena, metrologia, kalibracja ekspresji genów, standardy biomolekularne