Clear Sky Science · pl
Kontrola funkcji lizosomu przez białko aktywujące hydrolysis GTP TBC1D9B i jego partner wiążący TMEM55B
Jak centra recyklingu komórkowego utrzymują równowagę
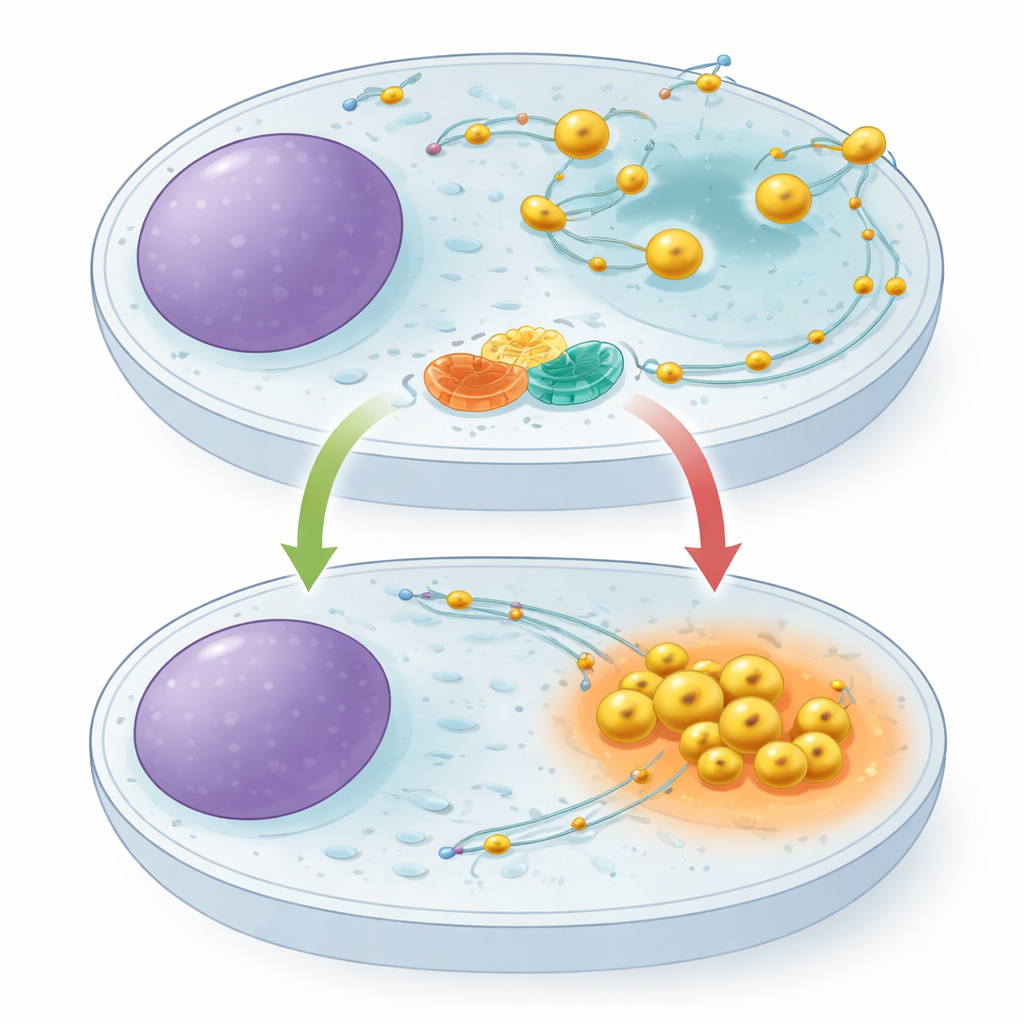
W każdej z naszych komórek znajdują się maleńkie pęcherzyki zwane lizosomami, które działają jak centra recyklingu i ośrodki kontroli wzrostu. Gdy funkcjonują prawidłowo, zużyte elementy są rozkładane, a ich składniki - ponownie wykorzystywane; gdy zawodzą, odpady się gromadzą, co może prowadzić do chorób, w tym neurodegeneracji i raka. Badanie to ujawnia, jak dwa mało znane białka pomagają przełączać lizosomy między przemieszczającym się stanem sprzyjającym wzrostowi a skupionym stanem sprzyjającym oczyszczaniu odpadów, rzucając światło na to, jak komórki dostosowują się do obfitości i głodu.
Przemieszczanie centrów recyklingu w komórce
Lizosomy nie są przytwierdzone na stałe. Poruszają się wzdłuż wewnętrznych szlaków, ciągnięte na zewnątrz przez jeden zestaw motorów molekularnych i odciągane do środka przez inny. Ich położenie w komórce ma znaczenie: gdy składników odżywczych jest pod dostatkiem, lizosomy rozprzestrzeniają się ku obwodowi komórki i wspierają sygnały wzrostowe; gdy brak pokarmu, cofają się ku centrum, stają się bardziej kwaśne i zwiększają rozkład materii komórkowej. Małe białko działające jak przełącznik, ARL8, jest znane z przemieszczenia lizosomów na zewnątrz, ale do tej pory nikt nie zidentyfikował dedykowanego hamulca, który by je powstrzymywał. Autorzy przypuszczali, że taki hamulec będzie kluczowy, aby komórki mogły szybko przełączyć się w tryb oszczędzania podczas głodzenia.
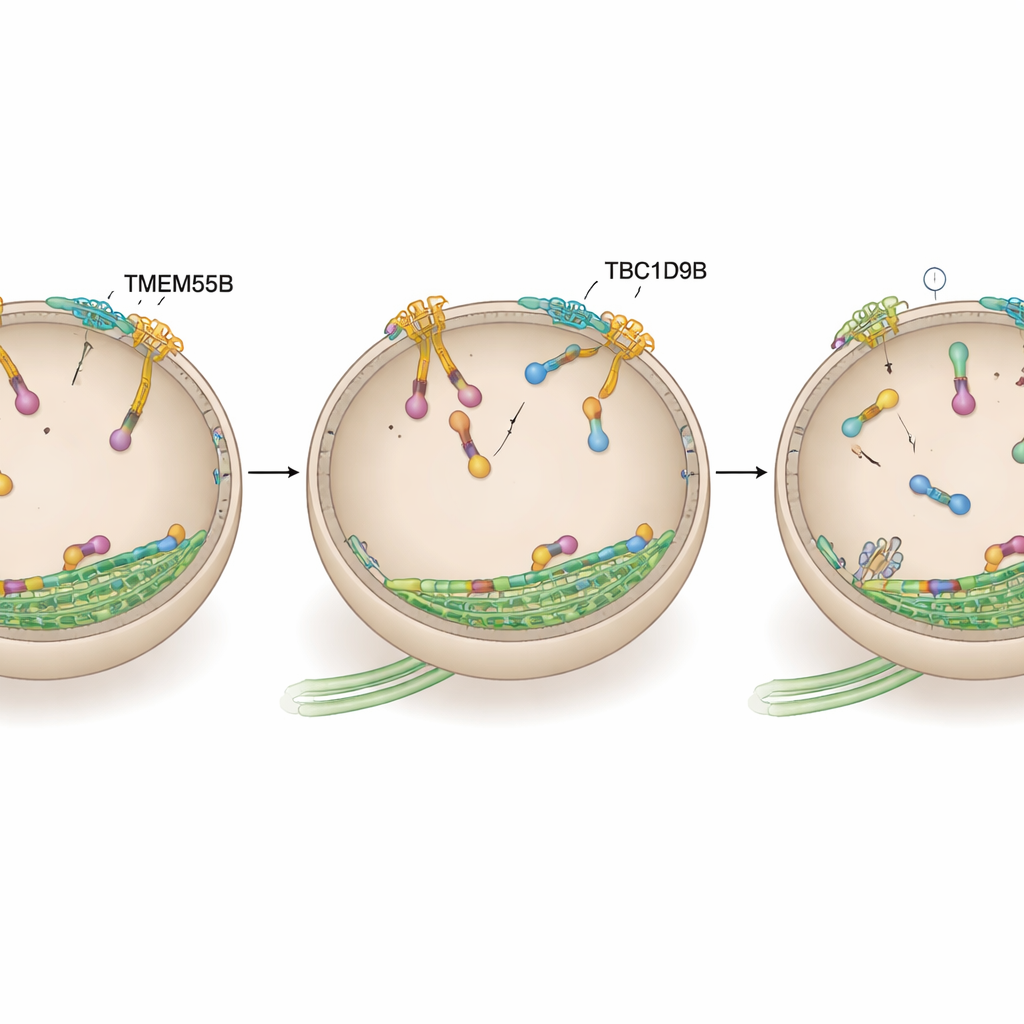
Molekularne partnerstwo na powierzchni lizosomu
Wykorzystując techniki „łowienia” białek i spektrometrię mas, badacze odkryli, że białko o nazwie TBC1D9B przyłącza się do błonowego białka lizosomu nazwanego TMEM55B. TMEM55B przebiega przez zewnętrzną powłokę lizosomu, podczas gdy TBC1D9B jest w dużej mierze rozpuszczalne i może dokować do błony. Zespół wykazał, że te dwa białka tworzą bezpośredni kompleks: oczyszczone TBC1D9B wiąże się z wystającą częścią TMEM55B w probówkach, a znakowane wersje tych białek wyciągają się wzajemnie z ekstraktów komórkowych. Dokładne mapowanie segmentów białkowych ujawniło, że kilka regionów TBC1D9B kontaktuje się z TMEM55B, lokując TBC1D9B na powierzchni lizosomu bez blokowania części białka odpowiedzialnej za jego aktywność katalityczną.
Gdy hamulec zawodzi, lizosomy szaleją
Aby sprawdzić, co robi to partnerstwo, autorzy użyli edycji CRISPR do usunięcia TMEM55B lub TBC1D9B z komórek ludzkich. W obu przypadkach lizosomy przestały się skupiać wokół jądra, a zamiast tego rozproszyły się ku krawędzi komórki i poruszały się szybciej wzdłuż swoich szlaków, naśladując stan ciągłego nasycenia. Ponowne wprowadzenie normalnego TBC1D9B przywróciło prawidłowe pozycjonowanie, ale wersja pozbawiona aktywności katalitycznej nie, co wskazuje, że jego aktywność enzymatyczna jest niezbędna. Gdy komórki były głodzone, kontrolne komórki ściągały lizosomy do wnętrza i zwiększały ich zdolności trawienne, co wykazano przez wzrost aktywności kluczowych enzymów i zwiększony rozkład testowego ładunku. Komórki pozbawione TMEM55B lub TBC1D9B nie potrafiły tego zrobić: ich lizosomy pozostały obwodowe, a ich odpowiedź degradacyjna na głodzenie była osłabiona. Autofagia, proces, w którym komórki „zjadają” uszkodzone elementy, także była upośledzona — mniej efektywnie przetwarzano reporter autofagii i następowało gromadzenie białka adaptorowego p62.
Wyłączanie silnika lizosomu
Zespół następnie zapytał, czy TBC1D9B wywiera swoje efekty poprzez bezpośrednie oddziaływanie z ARL8, przełącznikiem napędzającym ruch na zewnątrz. Używając znakowania bliskości w ludzkich neuronach oraz testów wiązania w liniach komórkowych i z oczyszczonymi białkami, wykazali, że TBC1D9B selektywnie wiąże aktywną, załadowaną GTP formę wariantu ARL8B, ale nie jego nieaktywną formę ani blisko spokrewnione ARL8A. Modele strukturalne przewidziały, że kluczowe reszty w TBC1D9B kontaktują kieszeń GTP ARL8B. W testach biochemicznych TBC1D9B przyspieszał hydrolizę GTP związanej z ARL8B, skutecznie przekształcając białko ze stanu „włączonego” w „wyłączony”; mutacyjna wersja TBC1D9B pozbawiona tych reszt nie potrafiła tego zrobić. Zgodnie z tym, komórki pozbawione TMEM55B lub TBC1D9B wykazywały zwiększoną ilość ARL8B na lizosomach, podczas gdy nadekspresja TBC1D9B cofała lizosomy ku centrum, podobnie jak osłabienie aktywności ARL8B.
Nowy regulator domowych porządków komórkowych
Na koniec autorzy sprawdzili, czy ten hamulec ARL8B wyjaśnia zmiany komórkowe obserwowane przy braku TBC1D9B. Gdy ARL8B został wyczerpany, lizosomy pozostawały skupione wokół jądra niezależnie od obecności TMEM55B czy TBC1D9B, a defekty autofagii spowodowane utratą TBC1D9B zostały w dużej mierze zniwelowane. Dane wspólnie wspierają model, w którym TMEM55B rekrutuje TBC1D9B do lizosomów, gdzie inaktywuje ARL8B i pozwala lizosomom przełączyć się z rozproszonego, wspierającego wzrost stanu na zcentralizowany, ukierunkowany na trawienie. Dla osób niebędących specjalistami oznacza to, że badanie odkryło ważne „pokretło”, którego komórki używają, by zdecydować, kiedy intensywniej się recyklingować — proces mający znaczenie dla zaburzeń związanych z gromadzeniem odpadów w mózgu, metabolizmu i raka.
Cytowanie: Duhay, V., Tian, M., Kosieradzka, K. et al. Control of lysosome function by the GTPase-activating protein TBC1D9B and its binding partner TMEM55B. Nat Commun 17, 2487 (2026). https://doi.org/10.1038/s41467-026-70345-y
Słowa kluczowe: lizosomy, autofagia, sygnalizacja komórkowa, transport organelli, neurodegeneracja