Clear Sky Science · pl
Przywrócenie wczesnej poporodowej dysregulacji synaptycznej ratuje degenerację neuronów ruchowych w modelu myszy rdzeniowo‑opuszkowego zaniku mięśni
Dlaczego drobne wczesne zmiany mogą mieć znaczenie dla późniejszej słabości mięśni
Rdzeniowo‑opuszkowy zanik mięśni (SBMA) to rzadka choroba dziedziczna, w której dorośli, zazwyczaj mężczyźni, stopniowo tracą siłę w kończynach, tułowiu i gardle. Objawy pojawiają się w średnim wieku, ale subtelne problemy zaczynają się znacznie wcześniej. W tym badaniu zadano zaskakujące pytanie: czy krótkie zdarzenia w pierwszych dniach po urodzeniu mogą po cichu przygotować grunt pod utratę komórek nerwowych dekady później — i czy skorygowanie tych wczesnych zaburzeń może chronić funkcję ruchową?
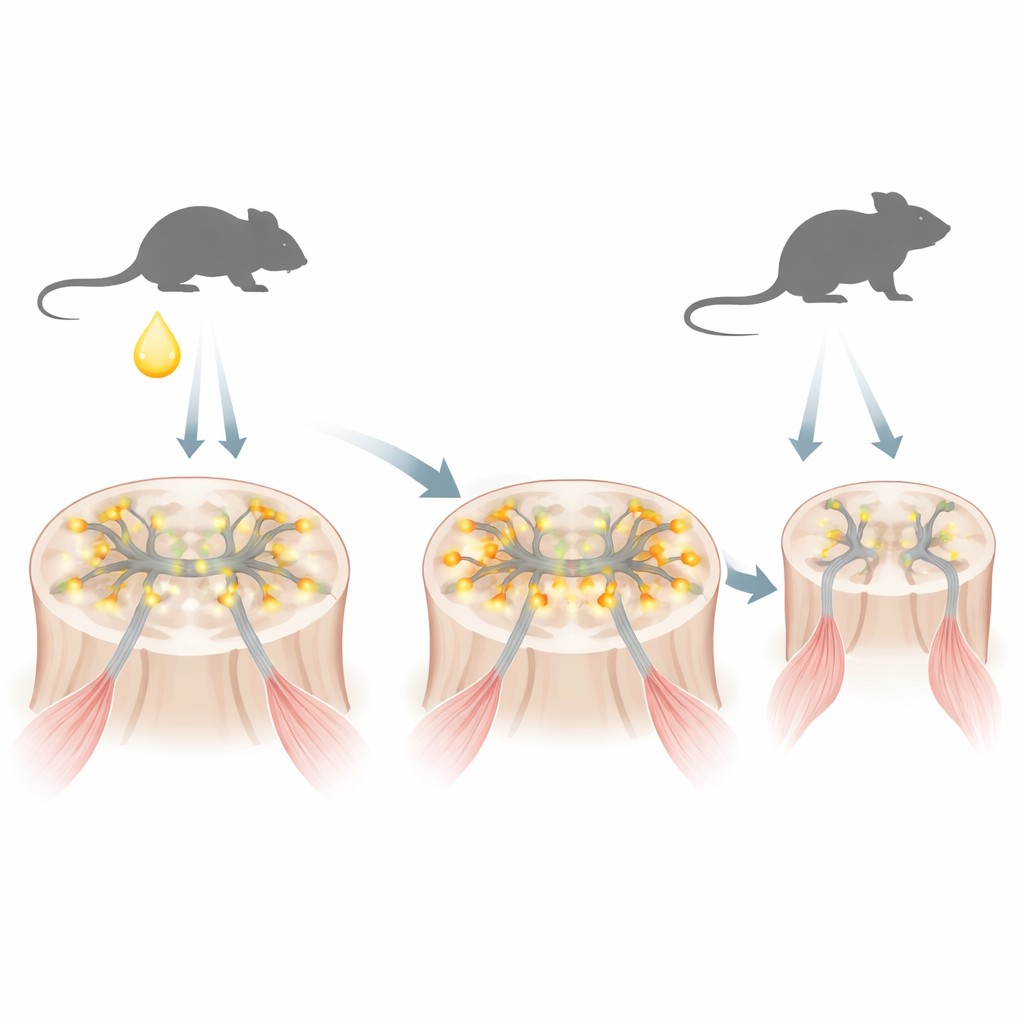
Choroba zakorzeniona w hormono‑wrażliwym przełączniku
SBMA jest spowodowany zmienioną wersją receptora androgenowego, białka wyczuwającego męskie hormony, takie jak testosteron. Zmodyfikowany receptor ma nadmiernie długi odcinek aminokwasu glutaminy. W modelu myszy odwzorowującym ludzką chorobę autorzy stwierdzili, że tuż po urodzeniu, gdy u samców krótko następuje wzrost testosteronu, mutantowy receptor szybko gromadzi się w jądrach neuronów ruchowych — komórek nerwowych kontrolujących mięśnie. Na tym wczesnym etapie białko jeszcze nie zlewa się w duże agregaty typowo związane z neurodegeneracją, ale już zmienia to, które geny są włączane i wyłączane.
Wczesne przeregulowanie synaps i niespokojne neurony ruchowe
Wykorzystując sekwencjonowanie RNA obejmujące cały genom w rdzeniach kręgowych nowo narodzonych myszy, zespół odkrył, że wiele genów związanych z synapsami pobudzającymi — punktami kontaktu, w których komórki nerwowe przekazują sygnały — było nadmiernie aktywnych. Wiele z tych genów koduje receptory glutaminianowe, które zwiększają skłonność neuronów do wyładowań. Grupę tę powiązano z zaburzeniem czynności REST, głównego „hamulca” białkowego, który normalnie utrzymuje takie geny synaptyczne pod ścisłą kontrolą podczas rozwoju. U myszy z SBMA i w neuronach ruchowych wyhodowanych z indukowanych komórek macierzystych pacjentów aktywność REST była osłabiona, a faworyzowana była skrócona forma zwana REST4, co zwalniało hamulec i zwiększało ekspresję genów synaps glutaminergicznych. Zgodnie z tym nowo narodzone neurony ruchowe SBMA wykazywały wyższe poziomy c‑Fos, markera niedawnej aktywności, a neurony pochodzące od pacjentów miały silniejsze i częstsze skoki wapniowe — cechy hiperaktywności.
Krótkie wczesne leczenie zmieniające bieg choroby na całe życie
Następnie badacze zapytali, czy zmniejszenie aktywności mutantowego receptora lub przywrócenie hamulca REST tylko w tym noworodkowym oknie mogłoby zmienić długoterminowy przebieg choroby. Podali oligonukleotydy antysensowne — krótkie nici zmodyfikowanego materiału genetycznego — do płynu otaczającego mózg i rdzeń kręgowy jednodniowych myszy SBMA. Jeden rodzaj oligonukleotydu przejściowo obniżał zarówno mutantowy, jak i normalny receptor androgenowy w ośrodkowym układzie nerwowym. Drugi rodzaj kierował składaniem mRNA REST z dala od REST4 z powrotem w stronę pełnej długości REST, ograniczając w ten sposób geny synaptyczne. Co zdumiewające, chociaż te terapie podano tylko raz, a ich bezpośrednie molekularne efekty wygasły w ciągu kilku tygodni, myszy żyły dłużej, lepiej chodziły po wirującym pręcie i zachowały silniejszy chwyt w późniejszym życiu. Ich neurony ruchowe i włókna mięśniowe były mniej zaniknięte, a wczesne markery nadaktywności neuronów i późniejsze wzrosty peptydów związanych ze stresem były osłabione.
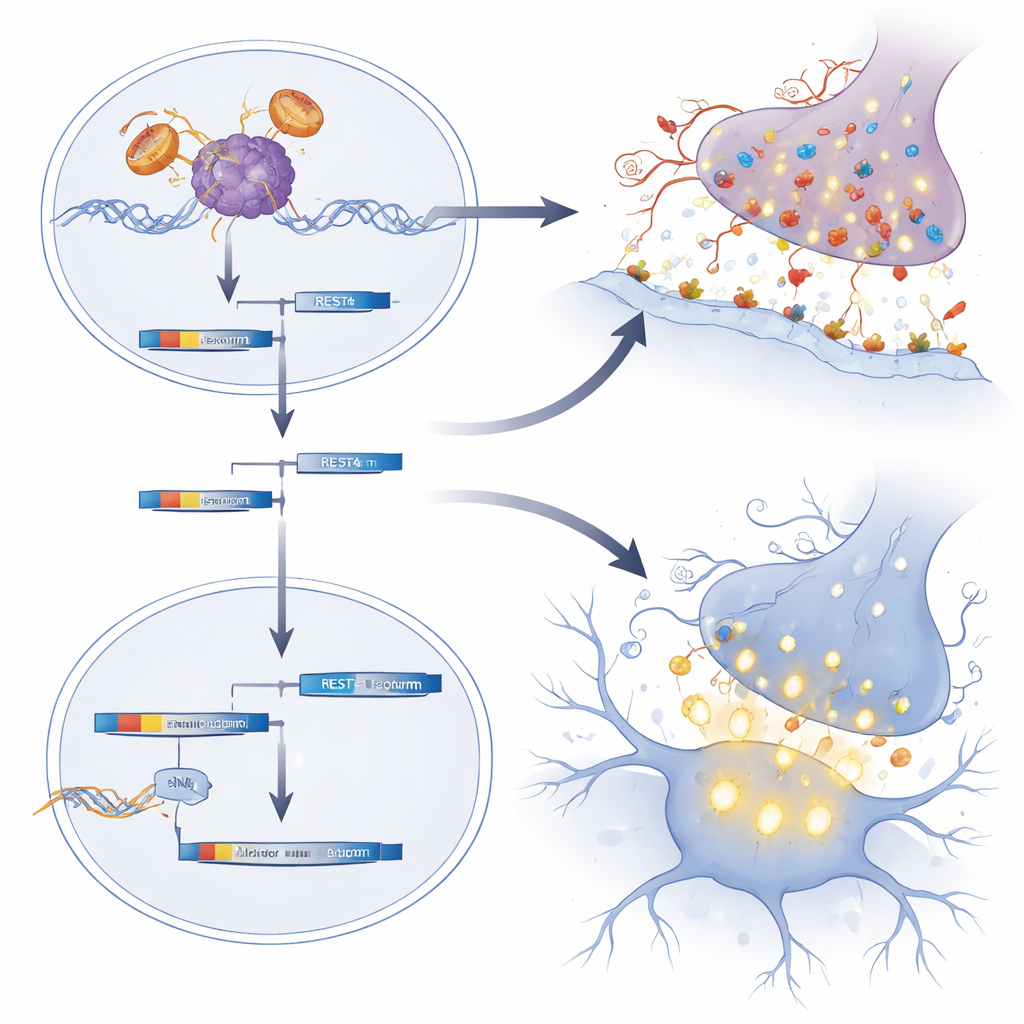
Jak wczesne hormony i strojenie genów kształtują podatność
Praca podkreśla także szczególną podatność neuronów ruchowych na krótkotrwały wyrzut testosteronu, który występuje wkrótce po urodzeniu. Gdy nowo narodzonym myszom SBMA podano dodatkowy testosteron, ich późniejsza słabość i utrata wagi uległy pogorszeniu, a programy genowe powiązane ze zdrową dojrzałością neuronów ruchowych zostały dodatkowo zaburzone. Normalne myszy nie wykazywały tych uszkodzeń, co podkreśla, że szkodliwy jest kombinowany efekt mutantowego receptora i hormonalnego wyrzutu. Razem wyniki sugerują, że w SBMA nadmiar synaps pobudzających i nadmiernie pobudliwe neurony ruchowe we wczesnym życiu stopniowo popychają układ ku awarii, mimo że oczywiste objawy nie pojawiają się aż do średniego wieku.
Co to oznacza dla osób żyjących z SBMA
Dla osoby niemającej specjalistycznej wiedzy kluczowe przesłanie jest takie, że SBMA może być częściowo chorobą wynikającą z nieprawidłowo zharmonizowanych i niewłaściwie okablowanych synaps w pierwszych dniach po urodzeniu. Wadliwy sensor hormonalny popycha rozwijające się neurony ruchowe w stan nadmiernego pobudzenia, a ten wczesny stres ostatecznie przyczynia się do ich degeneracji lata później. Zachęcające jest to, że precyzyjnie zaprojektowane leki genetyczne, podane w tych krytycznych oknach, mogą przywrócić równowagę sygnałów w neuronach ruchowych, uspokoić ich nadaktywność i znacząco opóźnić lub zmniejszyć późniejszą utratę komórek nerwowych u zwierząt. Choć przeniesienie takich interwencji z wczesnego okresu życia na ludzi będzie wymagało dużej ostrożności i dalszych badań, wyniki te wskazują na nowe strategie celujące u źródła SBMA na długo przed pojawieniem się osłabienia.
Cytowanie: Hirunagi, T., Sahashi, K., Iida, M. et al. Restoring early postnatal synaptic dysregulation rescues motor neuron degeneration in a mouse model of Spinal and Bulbar Muscular Atrophy. Nat Commun 17, 2412 (2026). https://doi.org/10.1038/s41467-026-70244-2
Słowa kluczowe: rdzeniowo‑opuszkowy zanik mięśni, hiperaktywność neuronów ruchowych, receptor androgenowy, regulacja synaptyczna REST, terapia oligonukleotydami antysensownymi