Clear Sky Science · pl
DiNovo umożliwia wysokie pokrycie i wysokie zaufanie w sekwencjonowaniu peptydów de novo dzięki enzymom lustrzanym i głębokiemu uczeniu
Nowe spojrzenie na białka
Białka to mikroskopijne maszyny podtrzymujące życie komórek, ale pełne odczytanie ich składników wciąż jest zaskakująco trudne. W artykule przedstawiono DiNovo, nowy system programowy, który pomaga naukowcom „czytać” fragmenty białek znacznie bardziej kompletnie i pewnie niż dotąd. Łącząc sprytne rozwiązanie biochemiczne z nowoczesną sztuczną inteligencją, narzędzie to obiecuje odkrywać ukryte białka, markery chorób, a nawet cele układu odpornościowego, które tradycyjne metody często pomijają.
Dlaczego odczytywanie fragmentów białek jest tak trudne
Większość współczesnych analiz białek polega na rozcinaniu ich na mniejsze części zwane peptydami, a następnie ważeniu fragmentów w spektrometrze mas. Na podstawie tych mas komputery próbują odtworzyć oryginalną sekwencję peptydu, jak rozwiązywanie krzyżówki na podstawie szczątkowych wskazówek. Istniejące metody zwykle zakładają, że peptydy pochodzą ze znanych baz białek, co dobrze działa dla dobrze poznanych białek, ale zawodzi dla nowych lub nieoczekiwanych. Tak zwane sekwencjonowanie de novo omija to ograniczenie, próbując odczytać peptydy bez odwołania do bazy, lecz często zawodzą, ponieważ brakuje niektórych fragmentów, a niektóre peptydy nigdy nie są czysto przycinane.
Wykorzystanie enzymów lustrzanych do uzupełniania braków
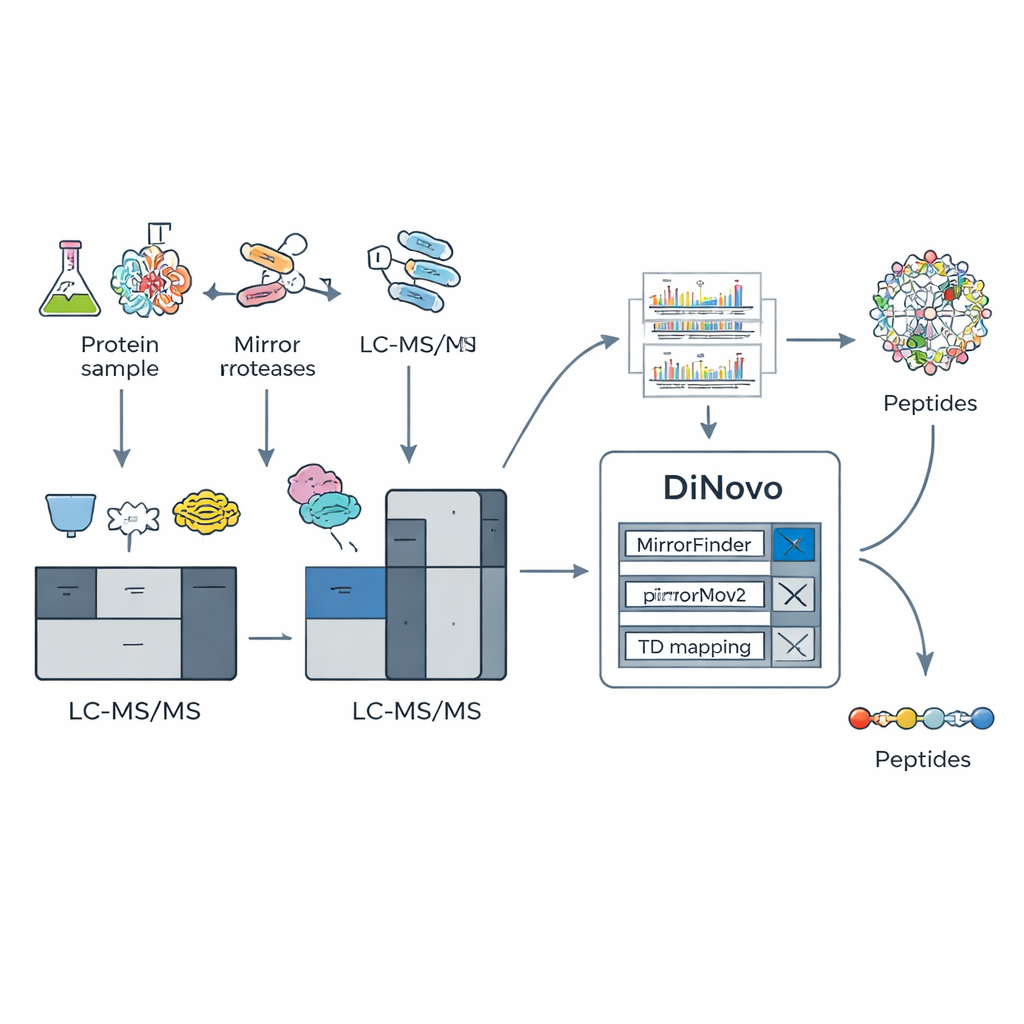
Kluczowa idea DiNovo polega na użyciu par „enzymów lustrzanych” – par enzymów tnących białka po przeciwnych stronach tego samego typu aminokwasu. Na przykład jeden enzym tnie tuż przed lizyną, podczas gdy jego partner tnie tuż po tej lizynie. Powstają w ten sposób dwa powiązane peptydy, które mają ten sam środkowy fragment, ale różne końce. Gdy te „lustrzane” peptydy są analizowane, ich widma mas zawierają komplementarne wzorce fragmentów: to, czego brakuje w jednym widmie, często pojawia się w drugim. Autorzy pokazują, że łączenie takich par lustrzanych może doprowadzić pokrycie fragmentów niemal do kompletnego, z około 98% możliwych cięć wspieranych przez rzeczywiste sygnały eksperymentalne, znacznie więcej niż przy użyciu jednego enzymu.
Inteligentny pipeline programowy stworzony dla danych lustrzanych
Aby wykorzystać ten trik biochemiczny, zespół zbudował DiNovo jako kompleksowy workflow programowy. Najpierw białka z bakterii i drożdży są trawione dwoma parami enzymów lustrzanych, a powstałe peptydy analizowane są wysokorozdzielczą spektrometrią mas. DiNovo wykorzystuje moduł zwany MirrorFinder do automatycznego rozpoznawania, które pary widm pochodzą od peptydów lustrzanych, robiąc to bezpośrednio na podstawie wzorców sygnału, a nie wcześniejszych przypuszczeń sekwencji. Następnie główny silnik de novo, MirrorNovo, używa głębokiego uczenia do interpretacji tych sparowanych widm, podczas gdy zapasowy, oparty na grafach silnik pNovoM2 oferuje szybszą opcję działającą tylko na CPU. Razem te narzędzia tłumaczą piki na sekwencje aminokwasów i analizują także pojedyncze widma, które nie utworzyły oczywistych par, wyciskając z danych jak najwięcej informacji.
Pomiary zaufania bez polegania na starych bazach
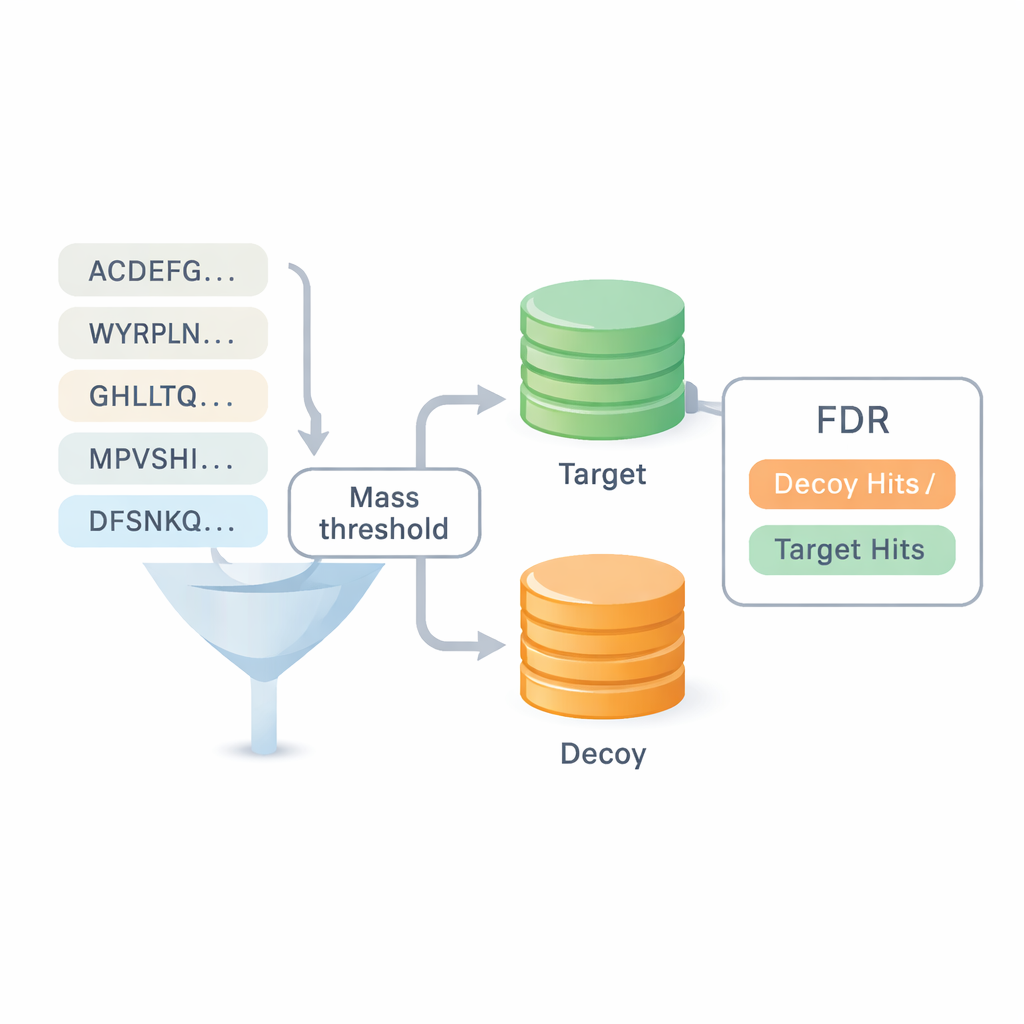
Jednym z największych pytań w sekwencjonowaniu de novo jest to, na ile ufać uzyskanym wynikom. Większość istniejących benchmarków wykorzystuje odpytania do baz danych, co zaciera granicę między podejściami i może ukrywać błędy. DiNovo wprowadza inną metodę kontroli jakości nazwaną mapowaniem cel–złudzenie (target-decoy mapping). Tutaj nowo odczytane peptydy mapowane są na połączony zbiór sekwencji białek prawdziwych (target) i sztucznych, przemieszanych (decoy). Porównując, jak często peptydy trafiają do rzeczywistego zbioru w porównaniu z przemieszanym, oprogramowanie może oszacować wskaźnik błędów, czyli false discovery rate, bez opierania się na wcześniejszych identyfikacjach. Pozwala to porównać DiNovo bezpośrednio z klasycznymi programami do przeszukiwania baz przy tych samych kryteriach błędu.
Co DiNovo dostarcza w praktyce
W testach na próbkach bakteryjnych, drożdżowych i przeciwciałowych, DiNovo konsekwentnie odczytywało znacznie więcej peptydów i aminokwasów niż znane narzędzia de novo używające tylko jednego enzymu. Używając dwóch par lustrzanych, wygenerowało 2–3 razy więcej aminokwasów o wysokim zaufaniu niż klasyczne ustawienie oparte tylko na trypsynie i zidentyfikowało więcej białek przy podobnym poziomie błędów. W porównaniu bezpośrednim z trzema wiodącymi silnikami przeszukiwania baz DiNovo znalazło podobne liczby aminokwasów i białek, a większość jego sekwencji zgadzała się z wynikami tych silników dla tych samych widm. Autorzy twierdzą, że ten poziom pokrycia i zgodności oznacza, iż sekwencjonowanie de novo, długo traktowane jako metoda awaryjna, może teraz stać obok przeszukiwań baz jako poważna, a w niektórych przypadkach lepsza, opcja.
Szeroki obraz: w kierunku pełnego, bezstronnego odczytu białek
Dla osoby niebędącej specjalistą kluczowa myśl jest taka, że DiNovo ułatwia znacznie dokładniejsze odczytywanie fragmentów białek bez ograniczeń wynikających z dostępnych baz referencyjnych. Podwajając lub potrajając ilość dobrze udokumentowanych informacji sekwencyjnych i dostarczając własne mechanizmy kontroli błędów, podejście to otwiera drzwi do odkrywania nieznanych białek, śledzenia subtelnych wariantów i badania złożonych mieszanin, w których wiele składników wciąż jest nieznanych. Krótko mówiąc, łącząc enzymy lustrzane z głębokim uczeniem i staranną statystyką, DiNovo pomaga przekształcić zaszumione ślady widmowe w wyraźniejszy, bardziej wiarygodny obraz białek leżących u podstaw zdrowia i chorób.
Cytowanie: Cao, Z., Peng, X., Zhang, D. et al. DiNovo enables high-coverage and high-confidence de novo peptide sequencing via mirror proteases and deep learning. Nat Commun 17, 2203 (2026). https://doi.org/10.1038/s41467-026-70224-6
Słowa kluczowe: proteomika, sekwencjonowanie peptydów de novo, spektrometria mas, głębokie uczenie, enzymy lustrzane