Clear Sky Science · pl
Wielo-modalna analiza komórkowo‑specyficznej patologii TDP-43 w korze ruchowej
Dlaczego te badania mają znaczenie dla ludzi
Stwardnienie zanikowe boczne (SLA) oraz otępienie czołowo‑skroniowe (FTD) to wyniszczające choroby mózgu, które odbierają ludziom ruch, mowę i osobowość. U większości chorych na SLA i u wielu pacjentów z FTD występuje wspólny mikroskopowy znak: gromadzenie się grudek białka TDP‑43 tam, gdzie nie powinno go być. Badanie stawia dwa praktyczne pytania o dużych implikacjach dla przyszłych terapii: które dokładnie komórki mózgu są najbardziej dotknięte zaburzeniami związanymi z TDP‑43 i co idzie nie tak w tych komórkach na poziomie regulacji DNA i aktywności genów?
Śledząc uszkodzenia w centrum kontroli ruchu
Naukowcy skupili się na pierwotnej korze ruchowej, pasku tkanki mózgowej kontrolującym ruchy dowolne. Wykorzystując pośmiertne próbki mózgu od osób z SLA, SLA‑FTD i od neurologicznie zdrowych kontrol, wyizolowali pojedyncze jądra komórkowe i odczytali zarówno aktywność genów, jak i stopień upakowania lokalnego DNA. Takie wielo‑omiczne podejście, zastosowane do ponad 180 000 jąder, pozwoliło im sklasyfikować komórki na precyzyjne typy: kilka klas neuronów pobudzających i hamujących oraz komórki wspierające, takie jak astrocyty, oligodendrocyty i mikroglej. Następnie połączyli to z przestrzennymi mapami ekspresji genów z innego ludzkiego zestawu danych, aby umieścić te typy komórek z powrotem w znajomej warstwowej strukturze kory.
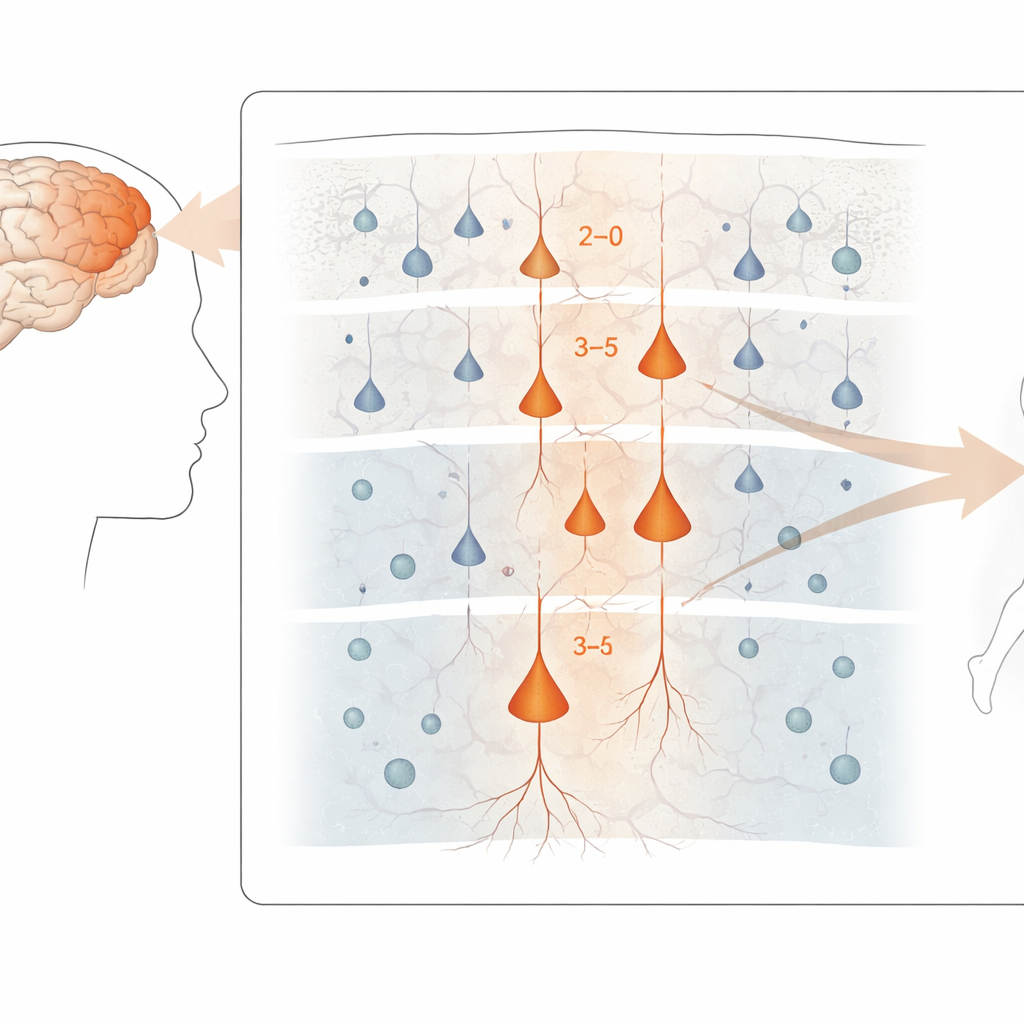
Wskazanie najbardziej wrażliwych neuronów
W całej korze ruchowej najsilniejsze zmiany związane z chorobą pojawiły się w neuronach pobudzających — komórkach napędzających aktywność w obrębie obwodów mózgowych. W szczególności neurony z górnych i środkowych warstw łączące się wewnątrz kory, a także niektóre komórki głębszych warstw wysyłające sygnały poza korę — w tym duże komórki „Betza”, kontrolujące neurony ruchowe rdzenia kręgowego — wykazywały najbardziej wyraźne zmiany. Dla porównania interneurony hamujące i wiele komórek glejowych było mniej dotkniętych na poziomie ekspresji genów, choć u niektórych z nich obserwowano delikatniejsze przemieszczenia. Pomimo tego molekularnego zamieszania, ogólny skład głównych typów komórek w tkance był zaskakująco podobny między pacjentami a kontrolami, co sugeruje, że uszkodzenie dotyczy bardziej funkcjonowania komórek niż jedynie ich utraty liczebnej.
Jak TDP-43 przekształca aktywność genów od środka
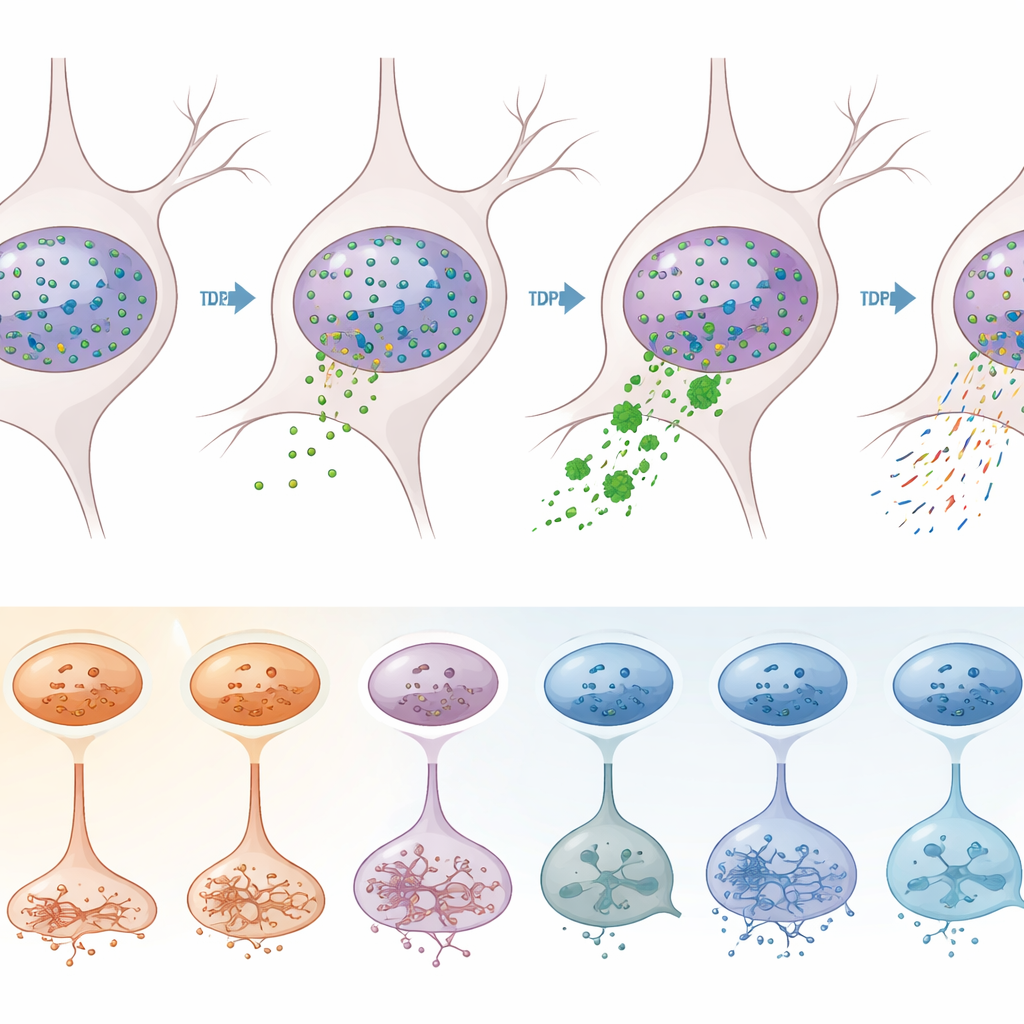
Aby oddzielić efekty bezpośrednio napędzane przez TDP‑43 od innych procesów chorobowych, zespół zastosował sprytną strategię sortowania. Oznakowali jądra przeciwciałami przeciw TDP‑43 i markerowi neuronalnemu, a następnie przy użyciu cytometrii przepływowej rozdzielili neurony, których jądra utraciły TDP‑43 (znak patologii) od tych, które go zachowały. Sekwencjonowanie ponad 12 000 takich jąder wykazało, że utrata TDP‑43 występuje w przeważającej mierze w neuronach pobudzających, szczególnie w określonych podtypach z warstw 2–3, 3–5, 5 i 6. W tych wrażliwych neuronach setki genów były źle regulowanych, w tym wiele już powiązanych z SLA. Klasyczne molekularne sygnatury dysfunkcji TDP‑43 — takie jak pojawienie się „kryptycznych” dodatkowych fragmentów w transkryptach genów STMN2 i KALRN oraz przesunięcia w miejscach cięcia i ogonkowania RNA — były wyraźnie wzbogacone w jądrach pozbawionych TDP‑43.
Remodeling epigenetyczny: nie wszystkie zmiany wynikają z TDP-43
Ponieważ mierzyli zarówno aktywność genów, jak i otwartość chromatyny w tych samych jądrach, autorzy mogli zapytać, które zmiany wiążą się ze zmianami upakowania DNA. Znaleźli dziesiątki tysięcy miejsc w genomie, gdzie lokalna dostępność chromatyny korelowała z ekspresją genów. Wiele z genów zmienionych w SLA i SLA‑FTD leżało w takich regionach, co wskazuje, że część sygnatury choroby odzwierciedla szerszy remodelings epigenetyczny, a nie bezpośrednie następstwo utraty TDP‑43. Co ciekawe, te związane z chromatyną zmiany często koncentrowały się na szlakach sygnałowych zaangażowanych w komunikację międzykomórkową i prowadzenie aksonów, i były szczególnie silne w niektórych neuronach pobudzających oraz oligodendrocytach. Porównując zmiany genowe powiązane z patologią TDP‑43 z tymi związanymi ze zmianami chromatyny, zespół stwierdził, że częściowo na siebie nachodzą, ale w dużej mierze stanowią odrębne warstwy zaburzeń.
Co to oznacza dla przyszłych terapii
Dla czytelnika niebędącego specjalistą kluczowy wniosek jest taki, że SLA i SLA‑FTD nie uszkadzają kory ruchowej równomiernie. Zamiast tego atakują konkretne typy neuronów pobudzających i w mniejszym stopniu niektóre komórki wspierające, zmieniając ich programy genowe w sposób zależny zarówno od zaburzeń funkcji TDP‑43, jak i od szerszych zmian w sposobie upakowania i odczytu DNA. Wyniki te sugerują, że skuteczne terapie mogą wymagać podejścia specyficznego dla typu komórki i konkretnego szlaku — na przykład przywrócenia funkcji TDP‑43 lub korekty jego błędów w składaniu (splicingu) w najbardziej wrażliwych neuronach, jednocześnie oddzielnie celując w zmiany epigenetyczne i sygnałowe wspólne dla wielu typów komórek. Mapując ten złożony krajobraz w wysokiej rozdzielczości, badanie dostarcza planu działania do projektowania bardziej precyzyjnych interwencji mających na celu spowolnienie lub zapobieganie utracie kontroli nad ruchem w SLA i SLA‑FTD.
Cytowanie: Ruf, W.P., Kühlwein, J.K., Meier, L. et al. Multi-modal dissection of cell-type specific TDP-43 pathology in the motor cortex. Nat Commun 17, 2406 (2026). https://doi.org/10.1038/s41467-026-69944-6
Słowa kluczowe: SLA, otępienie czołowo‑skroniowe, TDP-43, neurony kory ruchowej, single‑nucleus multiomics