Clear Sky Science · pl
Wydajne próbkowanie wielkoskalowych ścieżek przejściowych i pośrednich konformacji w sub-mezoskopowych kompleksach białkowych
Obserwowanie ruchu białek
Wiele cząsteczek niezbędnych do życia zachowuje się mniej jak sztywne klocki Lego, a bardziej jak maleńkie maszyny, które nieustannie zmieniają kształt. Te ruchy napędzają procesy takie jak wytwarzanie energii, naprawa DNA czy umożliwianie wirusom wejścia do komórek. Doświadczenia, takie jak krio‑elektronowa mikroskopia, potrafią teraz „zamrażać” niektóre z tych form, lecz nie ulotnych kroków pośrednich. W artykule przedstawiono eBDIMS2 — nową metodę komputerową, która potrafi „uzupełnić brakujące kadry” ruchu białek nawet dla olbrzymich maszyn molekularnych, które wcześniej były zbyt duże i złożone, by symulować je na zwykłym komputerze.
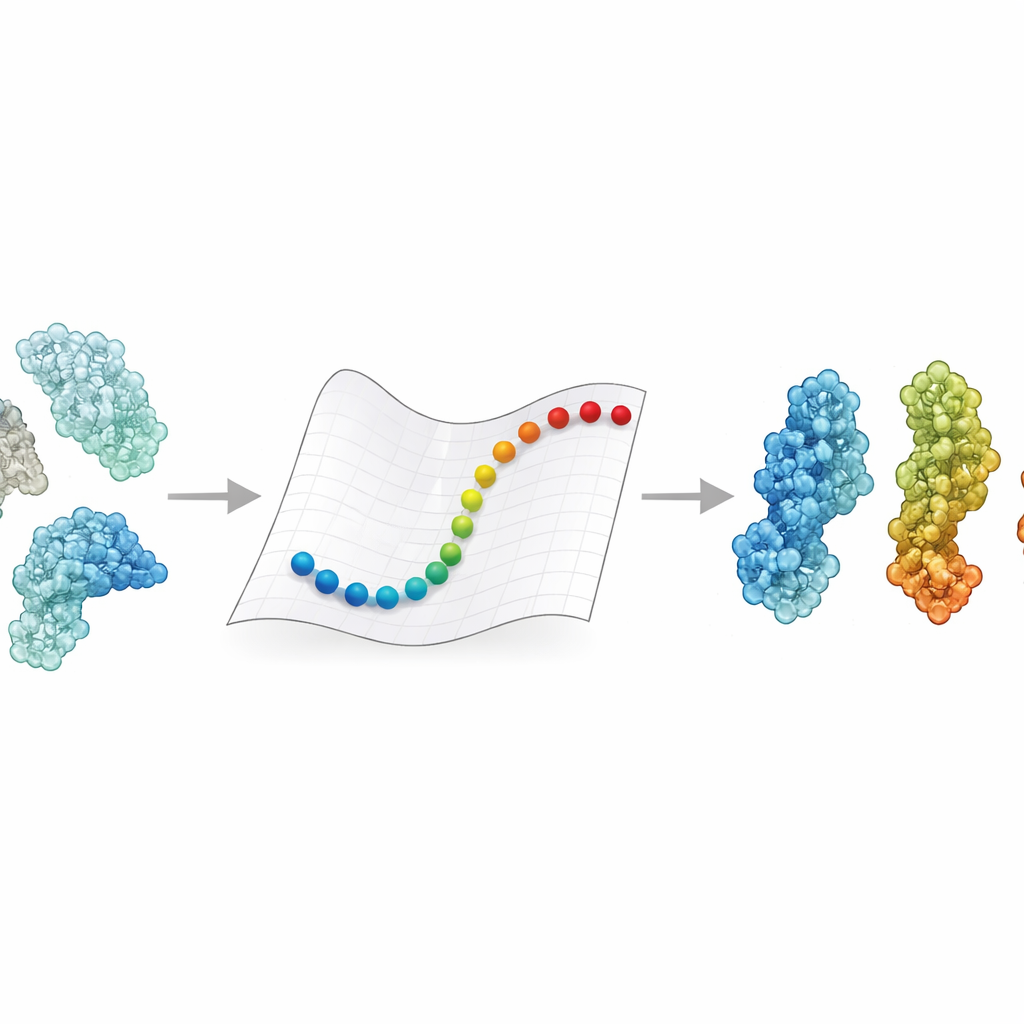
Dlaczego zmiany kształtu białek mają znaczenie
Białka rzadko pozostają w jednej, stałej pozycji. Otwierają się i zamykają, skręcają i wyginają w odpowiedzi na sygnały takie jak zmiany napięcia, pH czy wiązanie partnera. Te przemieszczenia mogą decydować o tym, czy enzym jest aktywny czy nie, albo czy receptor chwyci wirusa czy pozwoli mu umknąć. Eksperymenty dostarczają szczegółowych migawkowych obrazów kilku kluczowych form, a symulacje dynamiki molekularnej w zasadzie mogą je połączyć, śledząc każde atomowe położenie w czasie. Jednak śledzenie takich ruchów w przypadku ogromnych kompleksów widywanych teraz w krio‑EM, często ważących setki tysięcy do milionów Daltonów, zwykle wymaga superkomputerów i tygodni obliczeń. W efekcie dla wielu medycznie istotnych olbrzymów nadal nie wiemy, jak jedna forma przechodzi w drugą.
Szybsza trasa przez krajobrazy białkowe
eBDIMS2 stosuje skrót, upraszczając sposób reprezentacji białek i obliczania ich ruchu. Zamiast śledzić każdy atom, traktuje każdy aminokwas jako pojedynczy punkt połączony sprężynami w sieci elastycznej. Te sprężyny odzwierciedlają, jak różne fragmenty białka mają tendencję do współruchu. Metoda wykorzystuje następnie dynamikę Browna — reguły matematyczne naśladujące drgania w płynie — aby popchnąć strukturę z jednego doświadczalnie znanego stanu w kierunku drugiego. Kluczowe jest to, że eBDIMS2 skupia się tylko na istotnych oddziaływaniach, używając progów odległości i obliczeń równoległych, by obniżyć koszty. Poprawia to skalowanie programu z mniej więcej kwadratowego do prawie liniowego względem wielkości białka. W praktyce oznacza to, że przejścia dla ogromnych zespołów zbliżających się do dwóch milionów Daltonów można badać w ciągu godzin na komputerze stacjonarnym, zamiast być one praktycznie nieosiągalne.
Weryfikacja ścieżek na prawdziwych białkach
Aby sprawdzić, czy te szybkie ścieżki mają biologiczny sens, autorzy zgromadzili zespoły obejmujące 47 dużych białek i 15 dodatkowych kompleksów, łącznie setki struktur w większości rozwiązanych metodą krio‑elektronowej mikroskopii. Użyli analizy składowych głównych — narzędzia statystycznego wyodrębniającego dominujące sposoby ruchu każdego białka — aby uporządkować te struktury w krajobrazy konformacyjne, takie jak otwarte, zamknięte, aktywne czy nieaktywne. eBDIMS2 zostało poproszone o połączenie par stanów końcowych w tym krajobrazie. Powstałe ścieżki rzutowano z powrotem na te same mapy niskowymiarowe, aby ocenić, czy wyznaczają gładkie trasy przechodzące blisko eksperymentalnie obserwowanych pośredników. W ponad 30% systemów symulowane trasy przebiegały blisko — w odległości kilku angstremów — od pośrednich struktur, które nie były podawane jako dane wejściowe. W wymagających przypadkach, takich jak enzym naprawy DNA DNA‑PKcs czy białko kolca koronawirusa, ścieżki gruboziarniste dobrze pokrywały się także z dużo droższymi symulacjami na poziomie atomowym, w tym z ukierunkowaną dynamiką molekularną i zaawansowanymi metodami wzbogacania próbkowania.
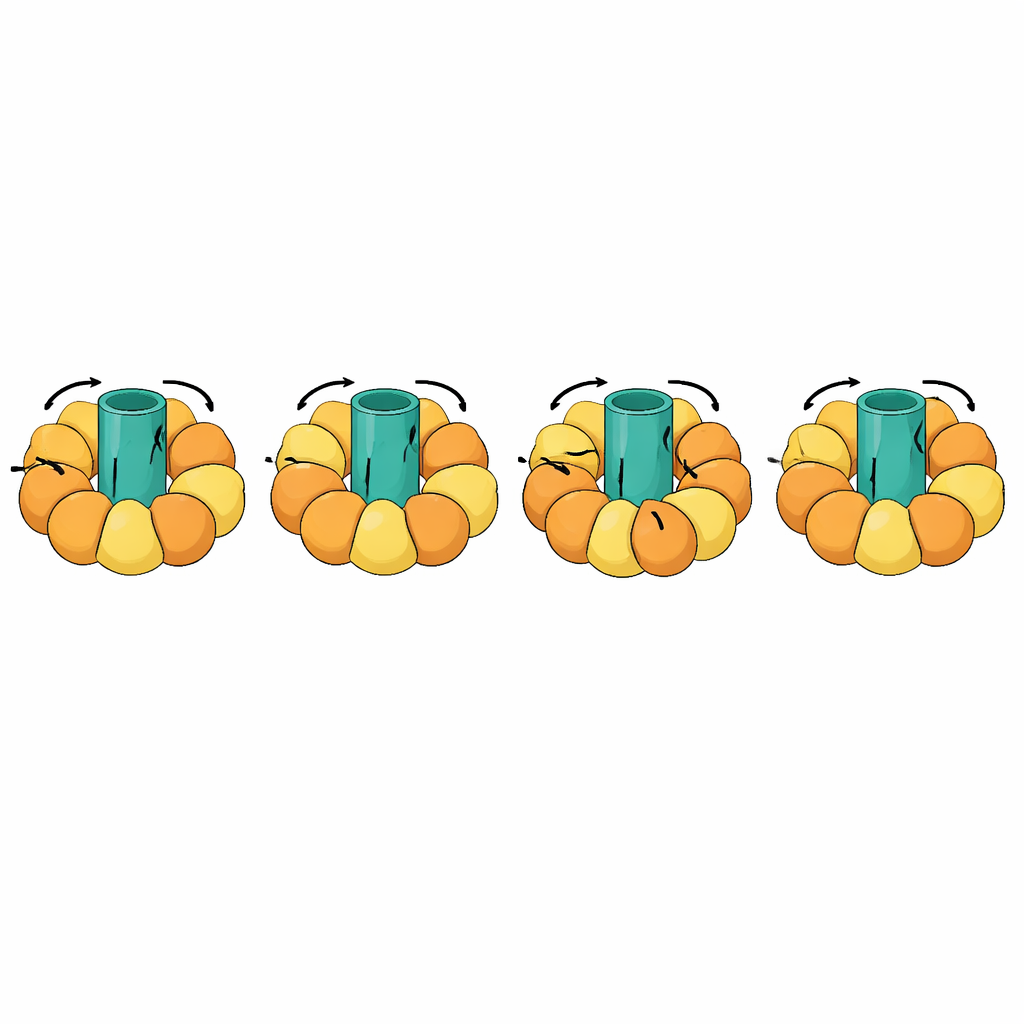
Śledzenie gigantycznych maszyn molekularnych
Jednym z najbardziej uderzających testów były maszyny obrotowe, takie jak syntazy ATP, które produkują energetyczną walutę komórkową przez sprzężenie wirującego rotora w błonie z otwieraniem i zamykaniem ruchów w otaczających podjednostkach. Te przejścia są wyjątkowo złożone: części cząsteczki muszą pozostać sztywne i obracać się jako całość, podczas gdy inne wyginają się w choreograficznym cyklu. eBDIMS2 wprowadza specjalne traktowanie dla takich quasi‑sztywnych fragmentów oraz dla niekompletnych modeli eksperymentalnych z brakującymi odcinkami — obu sytuacji częstych w krio‑EM. Dzięki tym funkcjom może symulować pełne cykle rotacyjne syntazy ATP i innych masywnych kompleksów, takich jak opiekuńcze białka chaperonowe, receptory czy zespoły wirusowe. W całym procesie wygenerowane struktury pośrednie unikają silnych zniekształceń, jakie dają niektóre konkurencyjne metody, i można je dopracować do modeli atomistycznych odpowiednich do obliczeń projektowania leków lub dłuższych, bardziej szczegółowych symulacji.
Co to oznacza dla biologii i medycyny
Badanie pokazuje, że eBDIMS2 potrafi wiarygodnie szkicować główne trasy między znanymi formami białek dla systemów, które były poza zasięgiem tradycyjnych symulacji. Nie zastępuje szczegółowych filmów na poziomie atomowym ani nie dostarcza precyzyjnych energii czy czasów, ale oferuje szybki, fizycznie ugruntowany sposób mapowania, jak wielkie maszyny molekularne mogą się poruszać, używając jedynie pary eksperymentalnych struktur jako danych wejściowych. W miarę jak bazy strukturalne zapełniają się wieloma stanami dużych zespołów białkowych powiązanych z rakiem, infekcjami i innymi chorobami, podejście to daje badaczom przystępne narzędzie do łączenia punktów, sugerowania wiarygodnych stanów pośrednich i wskazywania, gdzie warto sięgnąć po metody o wyższej rozdzielczości lub ukierunkowane projektowanie leków.
Cytowanie: Scaramozzino, D., Lee, B.H. & Orellana, L. Efficient sampling of large-scale transition pathways and intermediate conformations in sub-mesoscopic protein complexes. Nat Commun 17, 2202 (2026). https://doi.org/10.1038/s41467-026-69809-y
Słowa kluczowe: dynamika białek, symulacje molekularne, cryo-EM, ścieżki konformacyjne, modelowanie gruboziarniste