Clear Sky Science · pl
Aktywacja IRF3 w kardiomiocytach upośledza mitochondrialną funkcję oksydacyjną przez hamowanie PGC-1α i napędza niewydolność serca
Dlaczego zestresowane serca i zmęczone komórki mają znaczenie
Niewydolność serca często opisuje się jako „zużywanie się” serca, ale w istocie to także opowieść o chronicznym zapaleniu i wyczerpanych elektrowniach w komórkach mięśnia sercowego. W tym badaniu zadano pozornie proste pytanie o dużych implikacjach: czy istnieje pojedynczy molekularny przełącznik w komórkach serca, który łączy szkodliwe zapalenie z zawodzeniem produkcji energii — i jeśli tak, czy przełączenie go może zmienić przebieg niewydolności serca? Podążając tym wątkiem, autorzy odkrywają kluczowego gracza i pokazują, że delikatne wzmocnienie własnego programu energetycznego serca może częściowo uratować cierpiące serca myszy.
Molekularny przełącznik w chorych ludzkich sercach
Badacze skoncentrowali się na białku o nazwie IRF3, znanym przede wszystkim z roli w odpowiedzi komórkowej na infekcje wirusowe. Zbadali tkankę od osób z kardiomiopatią niedokrwienną, powszechną postacią niewydolności serca spowodowaną zmniejszonym przepływem krwi po zawale. W tych zawodzących sercach IRF3 był nie tylko obecny — był chemicznie aktywowany w określonych miejscach, co wskazywało, że aktywnie kieruje programami genowymi. Równocześnie mechanizmy pozwalające mitochondriom przekształcać paliwo w energię przez fosforylację oksydacyjną były wyraźnie osłabione. Podobny wzorzec zaobserwowano w modelach mysich z zawałem: po zaciśnięciu tętnicy wieńcowej IRF3 w komórkach mięśnia sercowego został silnie aktywowany, a geny kontrolowane przez IRF3 zostały uruchomione. Nawet fragmenty mitochondrialnego DNA — uwalniane z uszkodzonych mitochondriów i działające jako wewnętrzne sygnały „zagrożenia” — wystarczyły, by w izolowanych komórkach serca włączyć IRF3.
Wyłączenie IRF3 chroni serce
Aby sprawdzić, czy aktywność IRF3 w komórkach mięśnia sercowego rzeczywiście pogarsza chorobę, zespół stworzył myszy, w których IRF3 można było usunąć wyłącznie z kardiomiocytów, pozostawiając nietknięte pozostałe komórki odpornościowe i wspierające. Po wywołaniu zawału te myszy miały lepszą czynność wyrzutową i mniej blizn niż myszy normalne, mimo że początkowe uszkodzenie było takie samo. W komórkach serca hodowanych na szalce wygaszenie IRF3 osłabiło geny zapalne bez zakłócania innych powiązanych białek. Razem te wyniki przemawiają za tym, że IRF3 wewnątrz komórki sercowej nie jest tylko biernym obserwatorem: wzmacnia zapalenie i uszkodzenia strukturalne po niedokrwieniu i przyczynia się do przejścia do niewydolności serca.
Kiedy IRF3 jest „włączony” na stałe, system paliwowy zawodzi
Autorzy odwrócili następnie eksperyment: stworzyli myszy, w których IRF3 w kardiomiocytach można było wymusić w stanie trwale aktywnym za pomocą sprytnego genetycznego zabiegu „fosfomimetycznego”. Nawet bez zewnętrznego bodźca myszy te szybko rozwijały ciężką dysfunkcję serca, wysokie poziomy czynników zapalnych we krwi i oznaki uszkodzeń komórkowych. Głębsze badanie tkanki serca wykazało, że przy przewlekłej aktywności IRF3 tłumi on głównego koordynatora energetycznego zwanego PGC-1α. Ta cząsteczka zwykle sprzyja zdrowym mitochondriom, efektywnemu spalaniu tłuszczów i zrównoważonej gospodarce energetycznej komórki. Gdy PGC-1α jest obniżone, wiele białek mitochondrialnych spada, łańcuch transportu elektronów zawodza, a wybory paliwowe serca się zmieniają: spadają poziomy karnityny i związków związanych ze spalaniem tłuszczów, wykorzystanie ketonów jest zaburzone, a metabolizm glukozy ulega zniekształceniu. Nawet stosunek NAD⁺ do NADH — kluczowy wskaźnik równowagi redoks komórki — przechyla się w niekorzystnym kierunku.
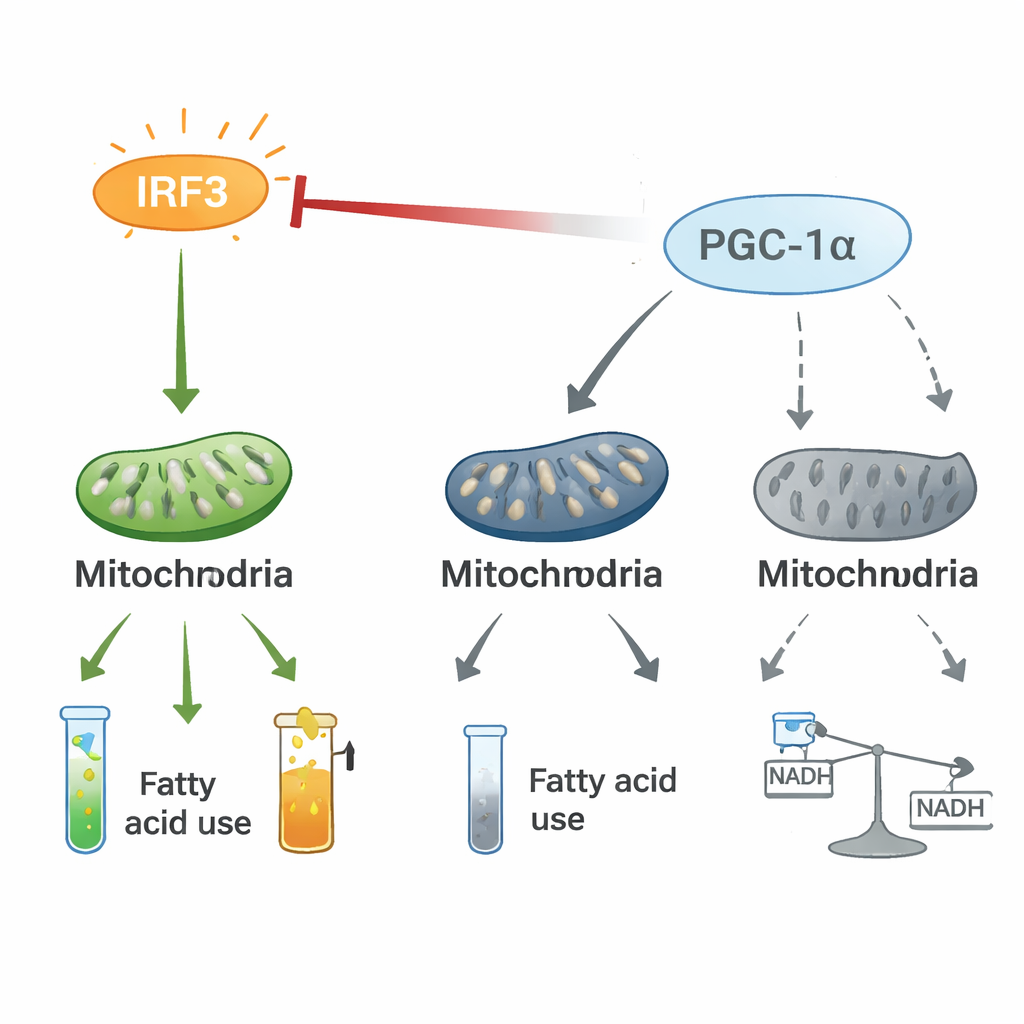
Sznur przeciągania między zapaleniem a kontrolą energii
Eksperymenty mechanistyczne ujawniły, że IRF3 i PGC-1α tworzą dwukierunkową oś regulacyjną. W komórkach serca aktywowany IRF3 fizycznie wiąże się z PGC-1α i osłabia jego zdolność do włączania genów spalania tłuszczów. Zmniejszenie poziomu IRF3 podnosi poziomy i aktywność PGC-1α, podczas gdy zwiększenie PGC-1α tłumi geny zapalne napędzane przez IRF3 i przywraca markery mitochondrialne, nawet w warunkach stresu, takich jak niedobór tlenu czy toksyny bakteryjne. Śledzenie izotopów stabilnych wykazało, że aktywacja IRF3 przekierowuje węgiel z normalnej produkcji energii przez cykl kwasu cytrynowego do alternatywnej ścieżki — szlaku pentozofosforanowego — i zaburza płynny przepływ metabolitów. Ten konflikt między prozapalnym przełącznikiem (IRF3) a współpilotem energetycznym (PGC-1α) wydaje się przekształcać metabolizm serca w sposób sprzyjający zapaleniu i utracie energii.
Delikatne doładowanie baterii serca
Wreszcie zespół zapytał, czy podniesienie PGC-1α może przeciwdziałać szkodom wywołanym przez IRF3. Użyli wektora terapii genowej ukierunkowanej na serce, aby umiarkowanie — lecz nie nadmiernie — zwiększyć PGC-1α w tych samych myszach z nadaktywowanym IRF3. Ten skromny wzrost poprawił czynność pompową, zwiększył białka mitochondrialne, wzmocnił geny związane ze spalaniem tłuszczów i metabolizmem NAD oraz zmniejszył aktywność genów zapalnych i fibrotcznych. W eksperymentach na komórkach jednoczesne wyrażenie PGC-1α z aktywnym IRF3 przywróciło zdrowszy stosunek NAD⁺/NADH i przywróciło korzystniejsze wykorzystanie paliw, skierowując je z powrotem w stronę tłuszczów. Dla czytelnika nieznającego szczegółów oznacza to, że ostrożne doładowanie „systemu zarządzania baterią” serca może częściowo skompensować szkodliwe skutki przewlekłego przełącznika zapalnego utkniętego w pozycji "włączone".
Co to oznacza dla przyszłej opieki nad niewydolnością serca
Ta praca stawia IRF3 jako centralne ogniwo łączące zapalenie i energetyczną niewydolność w komórkach mięśnia sercowego. Zamiast traktować zapalenie i metabolizm jako oddzielne problemy w niewydolności serca, badanie sugeruje, że są one powiązane osią IRF3–PGC-1α. Chociaż wyniki pochodzą z badań na myszach i komórkach, otwierają możliwość, że przyszłe terapie mogłyby albo wyciszać aktywność IRF3, albo wzmacniać PGC-1α i funkcję mitochondrialną, aby spowolnić lub zapobiec niewydolności serca po zawale. Mówiąc prosto: uspokojenie nadaktywnego wewnątrzkomórkowego systemu alarmowego i wsparcie elektrowni serca może okazać się silną, łączoną strategią, by dłużej utrzymać słabnące serca w dobrej kondycji.
Cytowanie: Kumari, M., Evangelakos, I., Deshpande, A. et al. Activation of IRF3 in cardiomyocytes impairs mitochondrial oxidative function through PGC-1α inhibition and drives heart failure. Nat Commun 17, 2051 (2026). https://doi.org/10.1038/s41467-026-69792-4
Słowa kluczowe: niewydolność serca, zapalenie, mitochondria, kardiomiocyty, PGC-1α