Clear Sky Science · pl
Sygnatury włączeń eksonów umożliwiają dokładne oszacowanie aktywności czynników splicingu
Odczytywanie ukrytych znaków edycji w komórce
Każda komórka w naszym ciele nieustannie edytuje swoje komunikaty RNA, zanim przekształci je w białka. Ta edycja, zwana splicingiem, pomaga zdecydować, czy komórka pozostanie zdrowa, czy przejdzie w stan nowotworowy. Badanie stojące za tym artykułem pokazuje, że analizując uważnie, które fragmenty RNA są zachowywane, a które pomijane — czyli sygnatury włączeń eksonów — naukowcy mogą z dużą precyzją wnioskować o aktywności molekularnych „redaktorów” kontrolujących splicing, nawet w skomplikowanych chorobach, takich jak rak.
Jak komórki wycinają i wklejają swoje komunikaty
Geny nie są odczytywane w jednym ciągłym kawałku. Zamiast tego komórki usuwają niekodujące fragmenty i zszywają ze sobą kodujące części, zwane eksonami, aby zbudować ostateczne komunikaty RNA. Specjalistyczne białka zwane czynnikami splicingu kierują tym procesem wycinania i wklejania, decydując, które eksony zostaną włączone. Ich zachowanie zależy od wielu warstw regulacji: ile własnego RNA i białka jest wytwarzane, jak są chemicznie modyfikowane, gdzie się znajdują w komórce i jak wchodzą w interakcje z innymi białkami. Ponieważ tak wiele dźwigni może zmieniać funkcję czynników splicingu, samo mierzenie jednego typu danych — na przykład ekspresji genów — często nie ujawnia, co te czynniki rzeczywiście robią.
Przekształcanie wzorców eksonów w odczyty aktywności
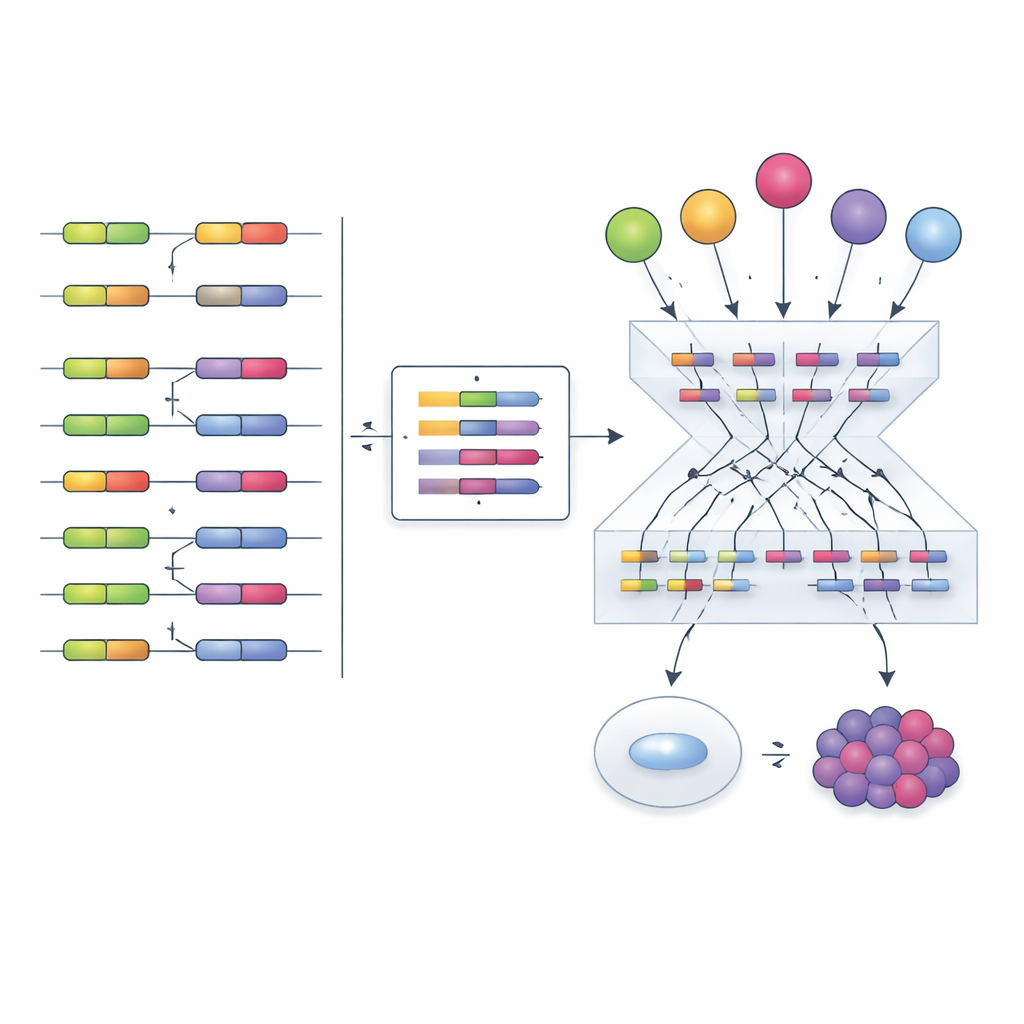
Zainspirowani wcześniejszymi pracami nad czynnikami transkrypcji, autorzy proponują inną strategię: zamiast próbować mierzyć czynniki splicingu bezpośrednio, odczytywać ich aktywność z efektów, jakie wywołują. Kiedy czynnik splicingu się zmienia, włączenie jego docelowych eksonów przesuwa się w rozpoznawalne wzorce. Zespół skompilował setki eksperymentów, w których pojedyncze czynniki splicingu były tłumione, usuwane lub nadmiernie eksprymowane, i użył tych danych do zbudowania „empirycznych sieci” łączących każdy czynnik z eksonami, które wyraźnie na niego reagują. Następnie zaadaptowali ramy obliczeniowe zwane VIPER, aby odczytać nową sygnaturę włączeń eksonów i ocenić, jak aktywny musiałby być każdy czynnik splicingu, by wyjaśnić obserwowany wzorzec.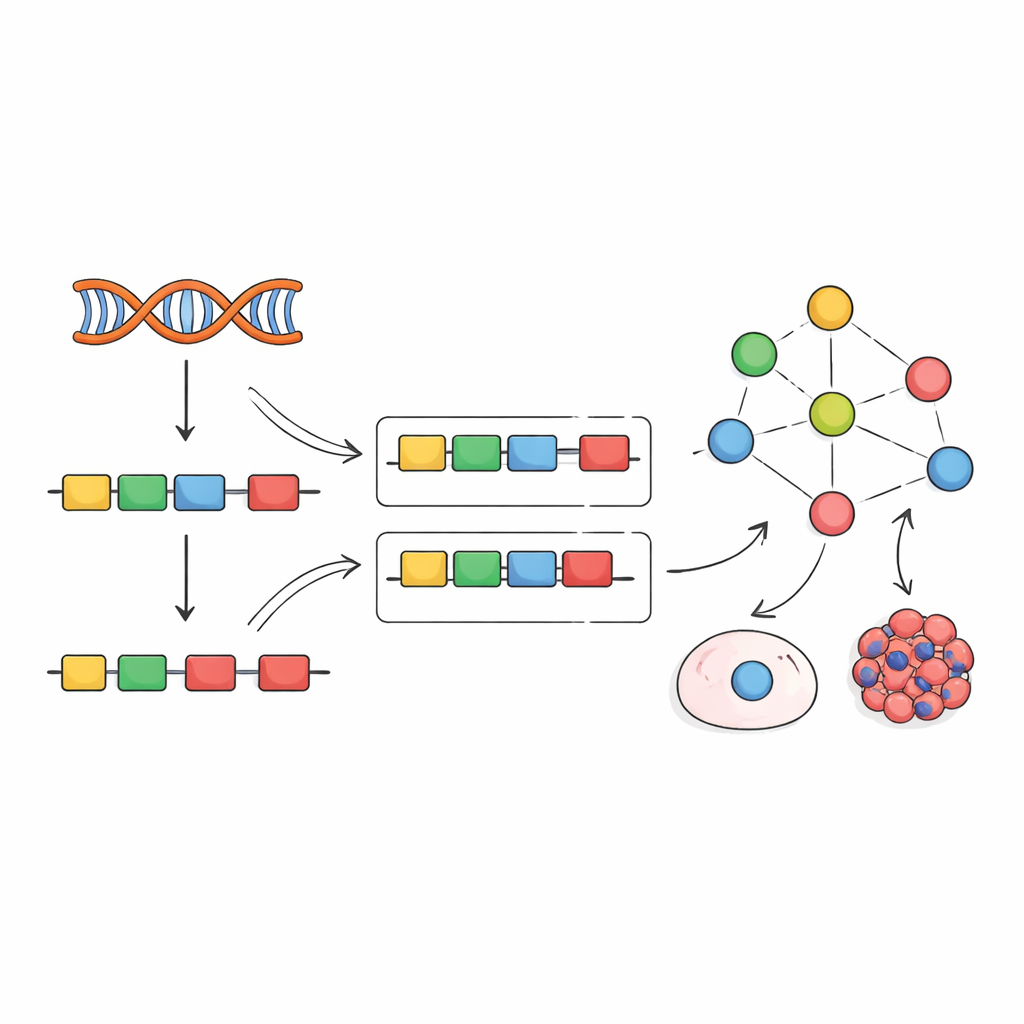
Testowanie metody na rzeczywistych zaburzeniach
Aby sprawdzić, czy podejście działa, badacze porównali kilka sposobów budowy sieci i obliczania wyników aktywności. Empiryczne sieci opracowane bezpośrednio na podstawie eksperymentów perturbacyjnych, w połączeniu z analizą wzbogacenia VIPER, wyraźnie przewyższały alternatywy oparte wyłącznie na wnioskowaniu statystycznym. Metoda poprawnie identyfikowała doświadczalnie zaburzony czynnik splicingu w większości testów, także między różnymi typami komórek i studiami. Ujmowała też subtelniejsze mechanizmy regulacyjne. Na przykład lek przeciwnowotworowy o nazwie Indisulam wywołuje degradację białka czynnika splicingu RBM39, podczas gdy poziomy jego RNA rosną w przypuszczalnym mechanizmie kompensacyjnym. Tradycyjna analiza ekspresji mylnie sugerowałaby większą aktywność RBM39, ale wynik oparty na eksonach prawidłowo ujawnił silną utratę funkcji, zgodną ze znanym mechanizmem działania leku.
Odkrywanie ukrytych programów splicingu w raku
Uzbrojeni w to narzędzie, autorzy sięgnęli po zasoby The Cancer Genome Atlas, analizując dane na poziomie eksonów z wielu typów nowotworów i odpowiadających im tkanek zdrowych. Odkryli dwa szerokie i powtarzalne programy splicingu. Jeden program obejmuje czynniki splicingu, które mają tendencję do wyższej aktywności w nowotworach i wiąże się z gorszym przeżyciem pacjentów — program przypominający onkogeniczny. Drugi zawiera czynniki systematycznie mniej aktywne w guzach i powiązane z lepszymi wynikami, przypominając programy supresorowe. Programy te obejmują geny zaangażowane w podstawowe cechy nowotworu, takie jak szybkie dzielenie komórek czy zdolność guzów do ukrywania się przed układem odpornościowym. Na przykład niektóre eksony regulowane przez program przypominający supresor nowotworu wydają się wpływać na to, jak dobrze pacjenci reagują na terapie hamujące punkty kontrolne układu odpornościowego, co wskazuje na nowe markery lub punkty interwencji.
Śledzenie zmian splicingu na drodze do raka
Zespół zbadał również model krok po kroku opisujący przejście komórek ludzkich od stanu normalnego do unieśmiertelnienia, formowania guza i w końcu przerzutowania. Stwierdzili, że program przypominający onkogeniczny staje się bardziej aktywny, gdy komórki nabywają mutacje napędzające raka, podczas gdy program przypominający supresor słabnie. Integrując wiele warstw danych — poziomy RNA, obfitość białek, modyfikacje chemiczne i zmiany splicingu wewnątrz samych czynników splicingu — zidentyfikowali skoncentrowany zestaw kandydatów na zdarzenia molekularne, które mogą napędzać te przesunięcia programowe, oferując priorytetyzowaną listę do przyszłych testów eksperymentalnych.
Dlaczego to ma znaczenie dla pacjentów i przyszłych badań
W istocie badanie pokazuje, że złożone zachowanie czynników splicingu można sprowadzić do jednego, interpretowalnego wyniku aktywności wyprowadzanego z tego, jak eksony są włączane lub pomijane. Umożliwia to badanie regulacji splicingu w dużych kohortach pacjentów i w różnych eksperymentach, używając jedynie standardowych danych z sekwencjonowania RNA, bez konieczności kosztownego profilowania multiomicznego. Dla czytelnika niebędącego specjalistą kluczowy przekaz jest taki, że wzorce w tym, jak geny są wycinane i składane, niosą bogate informacje o ukrytych systemach kontroli komórkowej, a odszyfrowanie tych wzorców może ujawnić nowe czynniki napędzające raka, poprawić prognozowanie i ukierunkować poszukiwanie bardziej precyzyjnych terapii.
Cytowanie: Anglada-Girotto, M., Segura-Morales, C., Moakley, D.F. et al. Exon inclusion signatures enable accurate estimation of splicing factor activity. Nat Commun 17, 1994 (2026). https://doi.org/10.1038/s41467-026-69642-3
Słowa kluczowe: montaż RNA, czynniki splicingowe, genomika nowotworów, transkryptomika, wnioskowanie aktywności białek