Clear Sky Science · pl
Nadmierne sygnalizowanie FGFR3 w achondroplazji zakłóca wymianę chondrocytów strefy spoczynkowej przez sygnalizację CREB
Dlaczego to badanie wzrostu kości ma znaczenie
Achondroplazja jest najczęstszą genetyczną przyczyną karłowatości z krótkimi kończynami. Dotyka nie tylko wzrostu, ale też zdrowia kręgosłupa, sprawności ruchowej i jakości życia. Obecne terapie pomagają, lecz nie przywracają w pełni wzrostu kości. W tym badaniu zastosowano zaawansowany model myszy, aby odkryć wcześniej pomijaną strefę problemową w rosnących kościach i wskazać nowy przełącznik sygnalizacyjny, zwany CREB, jako obiecujący cel dla przyszłych terapii.
Jak zdrowe kości wydłużają się
Długie kości, takie jak kość udowa, wydłużają się dzięki płytkom wzrostu u ich końców. Płytki wzrostu są zorganizowane w trzy główne warstwy komórek chrzęstnych. U góry znajduje się strefa spoczynkowa, gdzie komórki zachowują się jak pula komórek macierzystych, dzieląc się rzadko i wysyłając potomstwo w dół. Poniżej leży strefa proliferacyjna z szybko dzielącymi się komórkami ułożonymi w uporządkowane kolumny, które napędzają wydłużanie. Najdalej znajduje się strefa hipertroficzna z powiększonymi, dojrzałymi komórkami, które pomagają kierować tworzeniem nowej kości. Równowaga między tymi strefami utrzymuje prawidłową szybkość i kształt wzrostu kości.
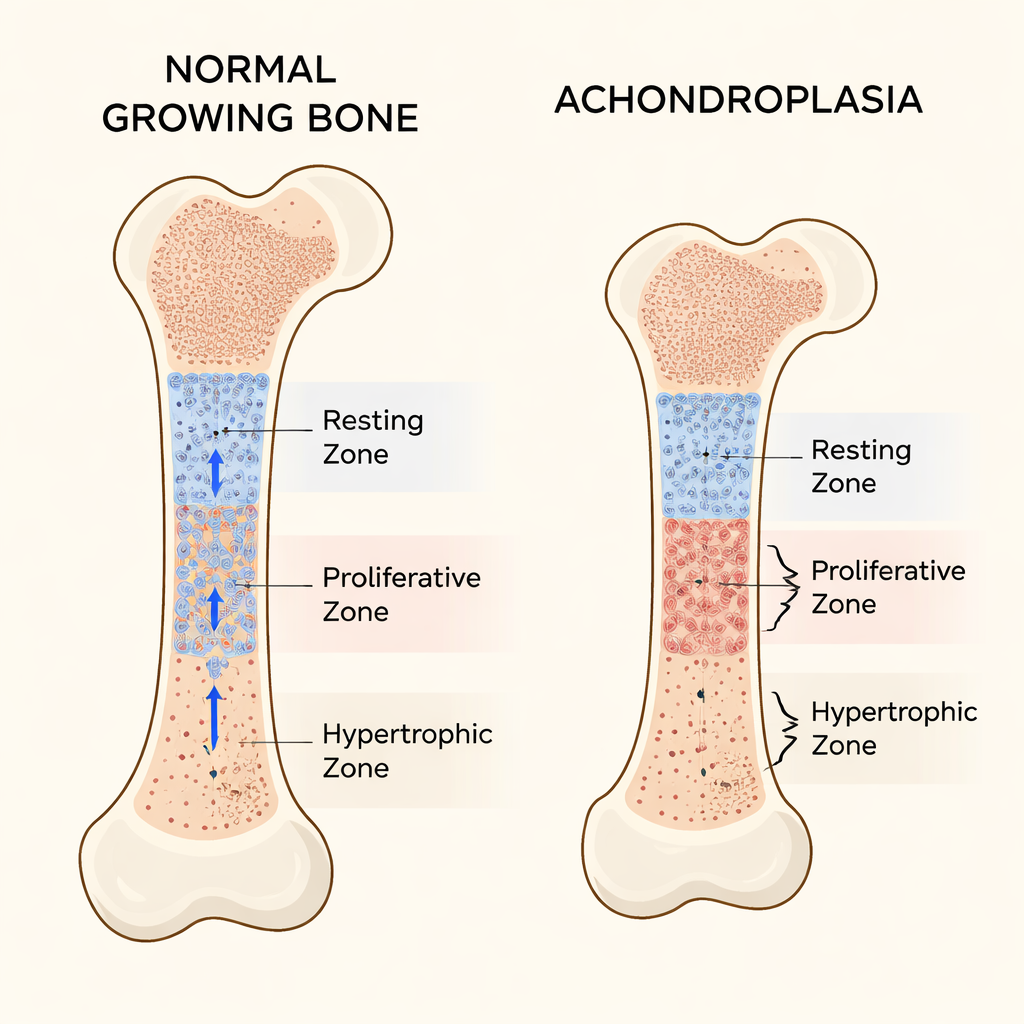
Co idzie nie tak w achondroplazji
U większości osób z achondroplazją pojedyncza mutacja w genie FGFR3 powoduje nadaktywność receptora, co hamuje wzrost kości. Wcześniejsze badania wykazały, że to sygnalizowanie spowalnia podziały komórek w strefie proliferacyjnej i blokuje końcowe powiększanie komórek w strefie hipertroficznej. Korzystając z myszy zaprojektowanych tak, by niosły ludzką mutację achondroplazji, autorzy potwierdzili silne skrócenie kończyn i płytek wzrostu. Dokładne pomiary ujawniły jednak coś wcześniej w dużej mierze pomijanego: sama strefa spoczynkowa stała się nietypowo gruba. Zamiast działać jako stałe, stabilne rezerwuar komórek macierzystych, obszar ten się rozszerzył i zawierał komórki ubogie w normalną macierz chrząstki.
Komórki strefy spoczynkowej tracą „cechy macierzyste”
Aby zrozumieć to rozszerzenie, zespół śledził, jak komórki płytki wzrostu dzieliły się i przemieszczały w czasie. U normalnych myszy komórki strefy spoczynkowej dzieliły się rzadko, a ich potomstwo migrowało w dół prostymi kolumnami, uzupełniając strefę proliferacyjną. U myszy mutantów znacznie więcej komórek w strefie spoczynkowej dzieliło się powoli i pozostawało na miejscu, tworząc zatłoczoną warstwę, która nie zasilała prawidłowo niższych stref. Śledzenie linii przy użyciu wielokolorowych etykiet genetycznych pokazało, że kolumny klonalne były krótkie i zdezorganizowane, a komórki potomne wędrowały w losowych kierunkach zamiast formować schludne stosy. Markery tożsamości przypominającej komórki macierzyste, takie jak białko CD73, zniknęły w powiększonej strefie spoczynkowej, co sugeruje, że nadaktywne FGFR3 zniszczyło normalną niszę komórek macierzystych.
Nowy podejrzany w sygnalizacji: CREB
Następnie badacze zastosowali sekwencjonowanie RNA pojedynczych komórek, aby profilować tysiące indywidualnych komórek płytki wzrostu. Zidentyfikowali odrębną grupę odpowiadającą rozszerzonej strefie spoczynkowej, bogatą między innymi w gen Spon1. Analiza szlaków wskazała na aktywację CREB, białka włączającego geny po przejściu w formę ufosforylowaną. Mikroskopia wykazała, że komórki strefy spoczynkowej u myszy mutantów silnie wykazywały aktywowany CREB i jego koaktywator CBP, wraz z wysokim poziomem FGFR3 i cząsteczek szlaku, takich jak STAT5. W hodowlach komórkowych pobudzenie szlaku FGFR3 zwiększało aktywność CREB i podnosiło poziom SPONDIN1 (białko SPON1), podczas gdy blokada FGFR3 lub CREB osłabiała te sygnały. To umieściło CREB jako kluczowy przekaźnik między nadaktywnym receptorem na powierzchni komórki a wadliwym zachowaniem komórek strefy spoczynkowej.
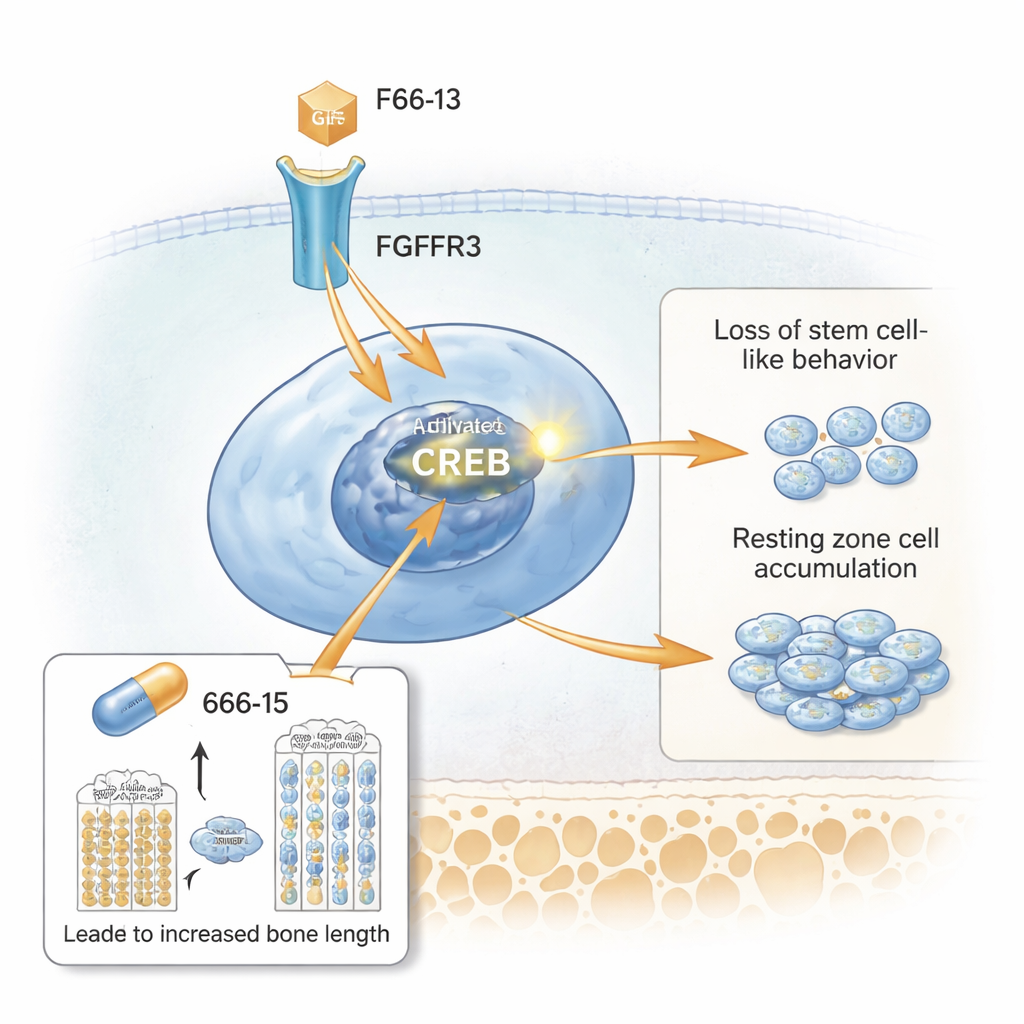
Testowanie leku tłumiącego CREB
Na koniec zespół sprawdził, czy wyciszenie CREB może złagodzić problemy wzrostowe. Leczyli myszy modelu achondroplazji małocząsteczkowym inhibitorem CREB, o nazwie 666-15, w okresie szybkiego wzrostu po porodzie. W porównaniu z nieleczonymi myszami mutantami, osobniki otrzymujące 666-15 miały większą masę ciała i dłuższe kości udowe. Ich płytki wzrostu wyglądały bardziej normalnie: strefa spoczynkowa się przerzedziła, strefy proliferacyjna i hipertroficzna odzyskały wysokość, a białka macierzy chrząstki pojawiły się ponownie. Markery nadaktywnej sygnalizacji CREB, w tym fosfo-CREB, SPONDIN1 i STAT5, zmalały w strefie spoczynkowej, podczas gdy marker przypominający komórkę macierzystą CD73 powrócił. Co ważne, ten sam lek przy zastosowanej dawce miał niewielki efekt u zdrowych myszy kontrolnych, co sugeruje, że działa głównie wtedy, gdy CREB jest nieprawidłowo podwyższony.
Co to oznacza dla przyszłych terapii
Badanie pokazuje, że w achondroplazji nadaktywne FGFR3 robi więcej niż tylko spowalnia podziały i powiększanie komórek; destabilizuje też cichą, przypominającą komórki macierzyste strefę spoczynkową przez włączenie CREB. To zaburzenie pozbawia niższe warstwy płytki wzrostu nowych komórek i przyczynia się do krótkich kości. Istniejące leki, takie jak vosoritide, celują głównie w inne ścieżki w strefach proliferacyjnej i hipertroficznej i tylko częściowo przywracają długość kości. Dodając CREB do listy celów — szczególnie w strefie spoczynkowej — przyszłe terapie skojarzone mogą lepiej normalizować wzrost u dzieci z achondroplazją.
Cytowanie: Horike, N., Oura, S., Koyamatsu, S. et al. Excess FGFR3 signaling in achondroplasia disrupts turnover of resting zone chondrocytes via CREB signaling. Nat Commun 17, 1856 (2026). https://doi.org/10.1038/s41467-026-69507-9
Słowa kluczowe: achondroplazja, FGFR3, płytka wzrostu, komórki macierzyste chrząstki, sygnalizacja CREB