Clear Sky Science · pl
XL-MSDigger: wszechstronne rozwiązanie oparte na głębokim uczeniu dla spektrometrii mas sprzężonej krzyżowo
Widzieć, jak białka trzymają się razem
Wszystkie procesy w naszych organizmach zależą nie tylko od prawidłowego fałdowania białek, lecz także od znajdowania przez nie właściwych partnerów. Obserwowanie tych molekularnych relacji w działaniu jest jednak niezwykle trudne. W pracy tej przedstawiono XL-MSDigger — platformę programową wykorzystującą nowoczesną sztuczną inteligencję do wydobywania znacznie wyraźniejszych sygnałów z hałaśliwej techniki eksperymentalnej zwanej spektrometrią mas sprzężoną krzyżowo, co pomaga naukowcom odwzorowywać, jak białka są zaaranżowane i z kim wchodzą w interakcje wewnątrz komórek.
Rozplątywanie zatłoczonego świata molekularnego
Aby dowiedzieć się, jak zbudowane są białka i jak się łączą, badacze często stosują spektrometrię mas sprzężoną krzyżowo. W tej metodzie małe chemiczne „mostki” łączą ze sobą pobliskie fragmenty białek. Połączone fragmenty są następnie rozbijane na fragmenty i ważone w spektrometrze mas. W zasadzie wzór fragmentów ujawnia, które części białka były blisko siebie w przestrzeni — jak znalezienie, które strony książki zostały spięte razem. W praktyce jednak dane są niezwykle złożone. Istniejące narzędzia komputerowe przetwarzają głównie podstawowe informacje o masie i mają trudności z ogromną liczbą możliwych kombinacji, co prowadzi do pomijania prawdziwych połączeń i generowania fałszywych trafień.
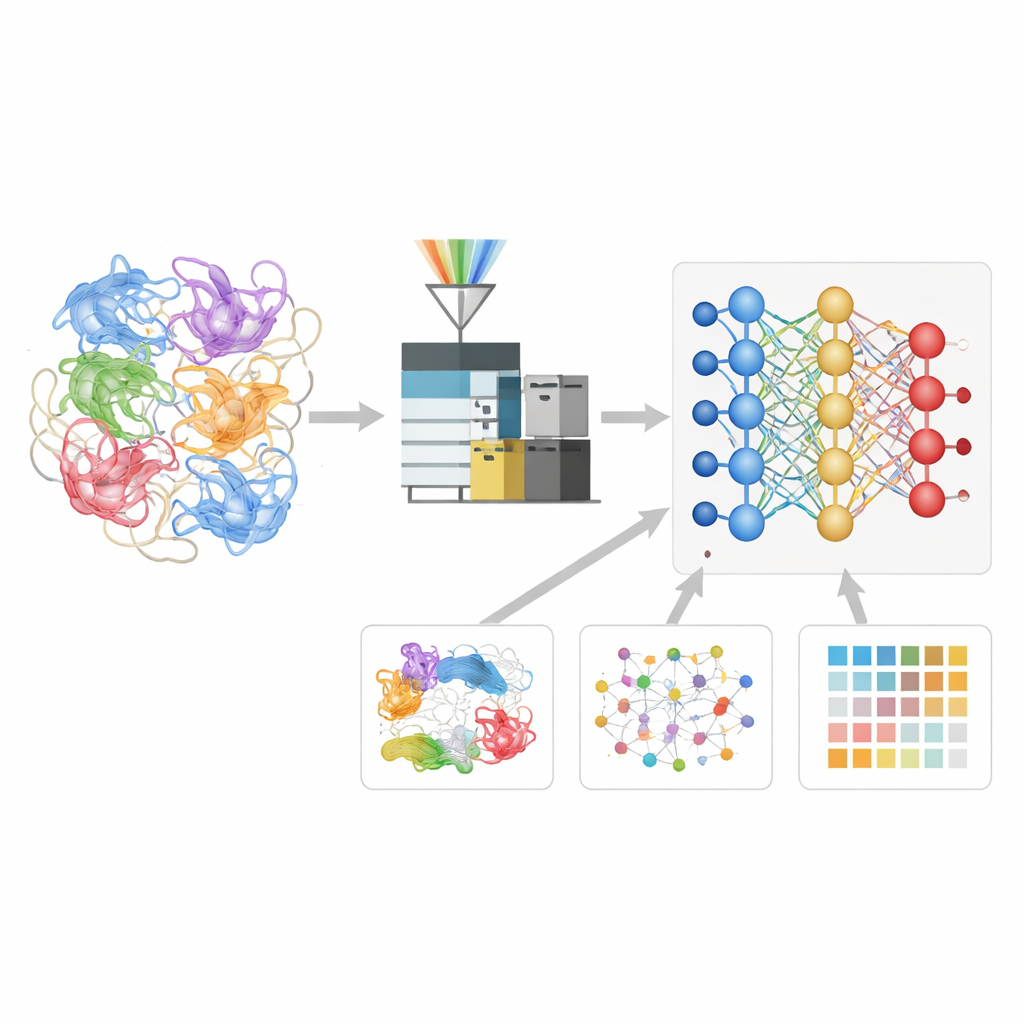
Nauczenie sieci neuronowej języka fragmentów białek
Autorzy stworzyli model głębokiego uczenia o nazwie Deep4D-XL, aby lepiej interpretować te eksperymenty krzyżowania. Najpierw zbudowali dużą bazę referencyjną, krzyżując białka z komórek ludzkich, rozbijając je na peptydy i rejestrując nie tylko ich masy, ale także czas przelotu przez instrument oraz zachowanie w komorze mobilności jonów. Każda para skrzyżowanych peptydów została zakodowana dla modelu, który wykorzystuje podwójną architekturę „syjamską” do jednoczesnego odczytu obu partnerów i krok cross-attention do łączenia ich informacji. Dzięki temu sieć uczy się przewidywać trzy kluczowe właściwości dowolnego nowego skrzyżowanego peptydu: kiedy powinien się pojawić w eksperymencie, jak powinien się przemieszczać i jak powinien wyglądać jego wzór fragmentacji.
Przekształcanie przewidywań w czystsze sygnały
XL-MSDigger opakowuje ten silnik predykcyjny w workflowy analityczne dla dwóch głównych trybów zbierania danych. W tradycyjnym, ukierunkowanym trybie instrument selektywnie rejestruje fragmenty jonów wybranych w locie. XL-MSDigger bierze początkowe dopasowania z ustalonych programów poszukujących i ponownie ocenia je, wykorzystując przewidywane zachowanie modelu dla każdego kandydata. Druga sieć neuronowa porównuje przewidywanie z eksperymentem w kilku wymiarach i przyznaje ulepszone oceny. Krok ponownego punktowania niemal podwaja liczbę pewnie wykrytych połączeń między różnymi białkami w próbkach drożdży i człowieka, przy utrzymaniu niskich wskaźników błędów, ujawniając znacznie więcej interakcji białko–białko niż wcześniej.
Rozumienie potoków nieukierunkowanych danych
Nowszy sposób pracy tych instrumentów, zwany akwizycją niezależną od danych, rejestruje fragmenty praktycznie wszystkiego w próbce, poprawiając pokrycie, ale generując przytłaczające ilości danych. Do tej pory nie było dobrego sposobu oszacowania, ile z uzyskanych skrzyżowań jest rzeczywiście prawdziwych. XL-MSDigger wykorzystuje Deep4D-XL do zbudowania starannie dopasowanej biblioteki „próbek” (decoy) fałszywych skrzyżowań, a następnie analizuje wpisy prawdziwe i decoy razem. Obserwując, jak często decoye przechodzą przez sito, oprogramowanie może oszacować współczynnik fałszywych odkryć i wytrenować kolejną sieć neuronową do rozdzielania prawdziwych i fałszywych dopasowań. To ponowne punktowanie zwiększa liczbę godnych zaufania sygnałów skrzyżowanych w przybliżeniu pięciokrotnie i daje wyraźne rozdzielenie między wzorcami prawdziwymi a decoy.
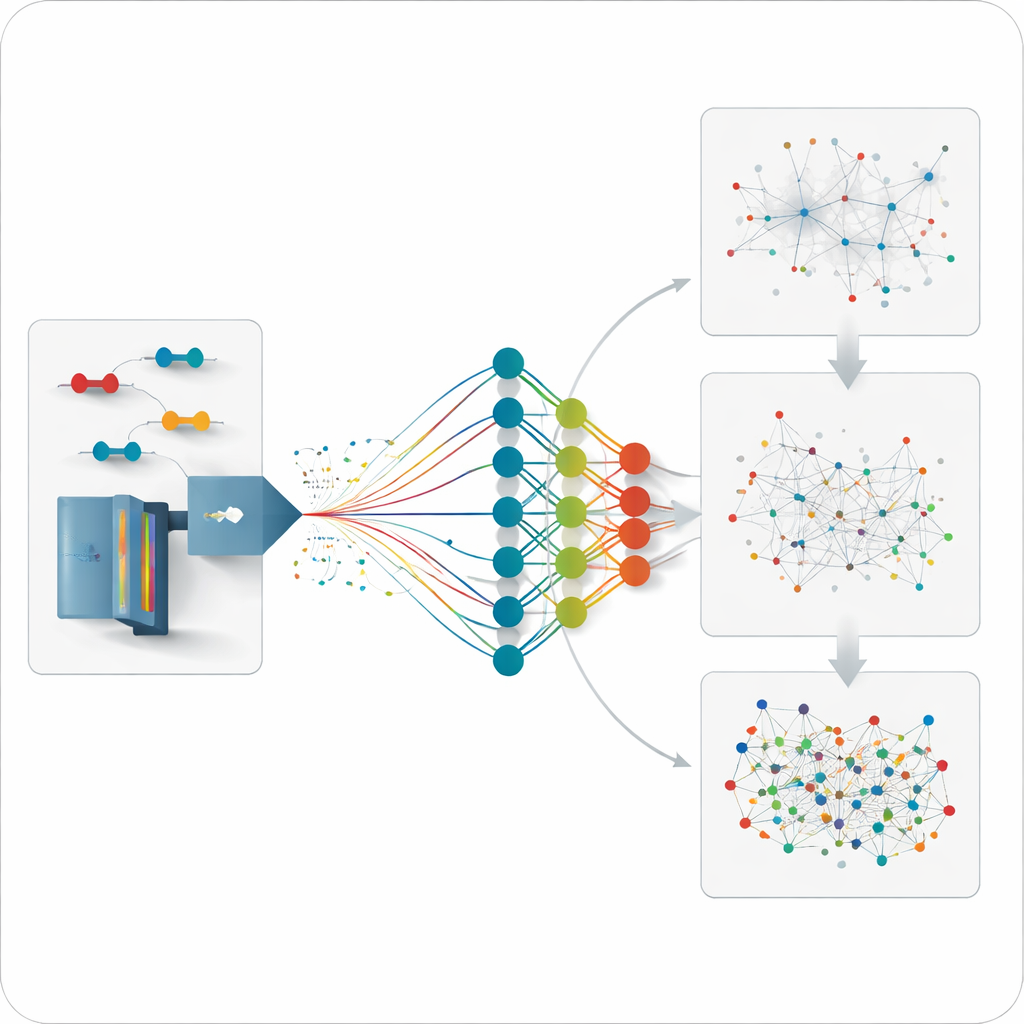
Przewidywanie tego, co jeszcze nie zostało zmierzone
Dzięki temu, że model potrafi prognozować zachowanie dowolnego prawdopodobnego skrzyżowanego peptydu, zespół może pójść o krok dalej i analizować dane dla połączeń, które nigdy wcześniej nie były bezpośrednio mierzone. Generują umiarkowanie duże, przewidywane biblioteki koncentrując się na wybranych białkach lub sieciach interakcji, a następnie przeszukują w stosunku do nich dane nieukierunkowane. Strategia ta odkrywa dodatkowe połączenia wewnątrz pojedynczych białek i między partnerami ważnych białek opiekuńczych (chaperonów), o odległościach dobrze zgodnych ze znanymi strukturami trójwymiarowymi. Odzyskuje też interakcje pominięte przez tradycyjne, bardziej ograniczone biblioteki eksperymentalne, zwłaszcza te o niskiej obfitości.
Otwarcie jaśniejszego okna na partnerstwa białek
Dla osób spoza specjalizacji kluczowa wiadomość jest taka, że XL-MSDigger działa jak wysoce wyszkolony rozpoznawacz wzorców nałożony na już potężną metodę eksperymentalną. Ucząc się, jak prawdziwe sygnały skrzyżowane powinny wyglądać jednocześnie w kilku wymiarach, potrafi przesiać ogromne, chaotyczne zbiory danych, odrzucić prawdopodobne podróbki i uratować prawdziwe, wcześniej ukryte połączenia białkowe. Choć pełne, całoproteomowe zastosowania nadal będą wymagać dużej mocy obliczeniowej, praca ta pokazuje, że połączenie eksperymentów krzyżowania z głębokim uczeniem może znacznie wyostrzyć nasz obraz rozmieszczenia białek i ich spotkań wewnątrz komórki.
Cytowanie: Chen, M., Hao, Y., Huang, X. et al. XL-MSDigger: a deep learning-based, versatile solution for cross-linking mass spectrometry. Nat Commun 17, 2554 (2026). https://doi.org/10.1038/s41467-026-69489-8
Słowa kluczowe: interakcje białek, spektrometria mas sprzężona krzyżowo, głębokie uczenie, proteomika, akwizycja niezależna od danych