Clear Sky Science · pl
Ogólny katalog referencyjny zdefiniowany przez szczyty konsensusu do analizy danych pojedynczych komórek ATAC-seq
Dlaczego mapowanie „otwartych drzwi” naszego DNA ma znaczenie
Każda komórka w twoim ciele zawiera w zasadzie tę samą sekwencję DNA, a jednak komórki mózgowe, krwi czy nowotworowe zachowują się bardzo odmiennie. Jednym z kluczowych powodów jest to, że tylko niektóre odcinki DNA są w danym momencie odsłonięte i „otwarte” do wykorzystania. Nowe technologie pojedynczych komórek potrafią teraz mierzyć tę otwartość w skali całego genomu, lecz do tej pory brakowało wspólnej mapy referencyjnej — czegoś na kształt standardowego atlasu — umożliwiającej porównania między eksperymentami i laboratoriami. W tym badaniu zbudowano taką mapę, nazwaną cPeaks, i pokazano, jak może ona wyostrzyć nasze rozumienie typów komórek, rozwoju i nowotworów.
Przekształcanie wielu eksperymentów w jedną wspólną mapę
Autorzy zaczęli od zgromadzenia 624 wysokiej jakości eksperymentów mierzących otwartą chromatynę — dostępne fragmenty DNA — obejmujących ponad 40 ludzkich narządów. W każdym eksperymencie programy komputerowe wcześniej zaznaczyły „szczyty”, gdzie DNA było szczególnie odsłonięte. Zamiast traktować każdy zbiór danych osobno, zespół starannie nałożył wszystkie te listy szczytów na genom i scalił zachodzące na siebie regiony. Następnie zbadali, jak często każda maleńka pozycja wewnątrz tych scalonych regionów była zgłaszana jako otwarta w różnych eksperymentach, zamieniając każdy region w charakterystyczny kształt odzwierciedlający, jak konsekwentnie się pojawiał. Gdy scalony region zawierał kilka blisko położonych miejsc otwartych, rozdzielili go na kilka prostszych jednostek. Te jednostki — około 1,4 miliona łącznie — stały się zaobserwowanymi szczytami konsensusu, czyli cPeaks, proponowanym katalogiem referencyjnym dostępności chromatyny u ludzi. 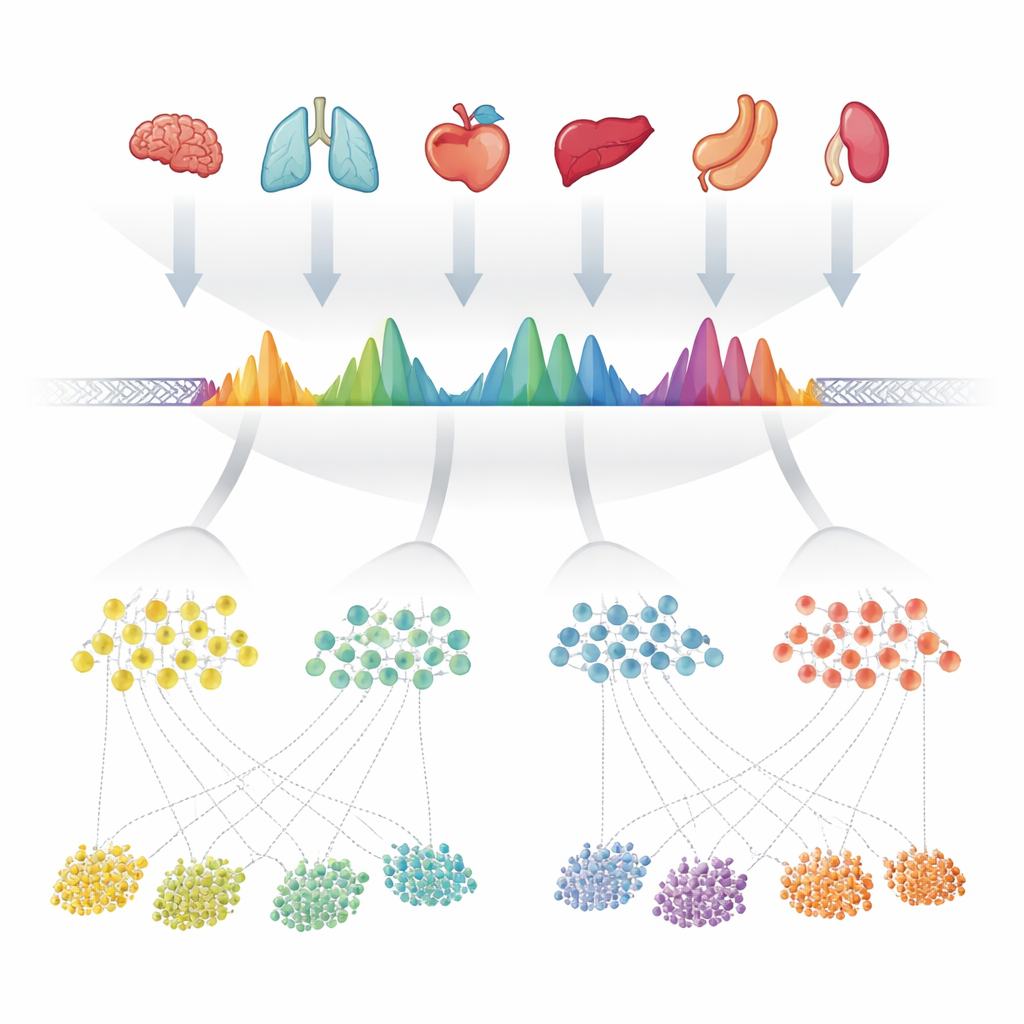
Stabilny odcisk palca w różnych tkankach i technologiach
Aby pełnić rolę użytecznej referencji, cPeaks muszą odzwierciedlać autentyczne, powtarzalne cechy genomu, a nie przypadłości konkretnych próbek czy oprogramowania. Autorzy sprawdzili to, rekonstruując swoje scalone regiony używając tylko próbek krwi, tylko tkanek litych, oddzielnych publicznych baz danych, a nawet różnych metod laboratoryjnych badających otwarte DNA. W każdym przypadku te same miejsca genomowe dawały zadziwiająco podobne kształty szczytów, a większość zestawów danych single-cell, które analizowali, pokrywała ponad 90% własnych szczytów z katalogiem cPeaks. Odczyty z wielu narządów gromadziły się precyzyjnie wokół środków cPeaks, co pokazuje, że te regiony wiarygodnie uchwytują miejsca otwartej chromatyny. W porównaniu z wcześniejszymi zestawami referencyjnymi opartymi na pokrewnych technologiach, cPeaks obejmowały większą część dostępnego DNA wykrywanego przez eksperymenty ATAC-seq i uchwyciły niemal tyle samo sygnału, co szczyty definiowane od podstaw w każdym zbiorze danych — mimo że są stałe i wielokrotnego użytku.
Nauczanie sieci neuronowej odnajdywania brakujących regionów
Nawet setki istniejących próbek nie obejmują wszystkich możliwych typów komórek. Aby rozszerzyć swoją mapę o regiony jeszcze niezaobserwowane, zespół zwrócił się ku głębokiemu uczeniu. Wytrenowali jednowymiarową splotową sieć neuronową na sekwencjach DNA: przykłady leżące w ramach zaobserwowanych cPeaks posłużyły jako pozytywy, a losowo wybrane regiony tła jako negatywy. Model nauczył się rozróżniać te dwie klasy z wysoką precyzją, co sugeruje, że cPeaks niosą rozpoznawalne wzorce sekwencyjne. Kiedy badacze celowo ukrywali szczyty specyficzne dla danej tkanki, sieć nadal potrafiła je odzyskać tylko na podstawie sekwencji, także rzadkie, tkankowo-specyficzne miejsca. Następnie przesunęli małe okno wzdłuż reszty genomu, punktując każdy segment i dodając około 280 000 wysoko punktowanych nowych regionów do katalogu jako przewidywane cPeaks, poprawiając szczególnie pokrycie tkanek niedostatecznie reprezentowanych w danych źródłowych.
Łączenie otwartych regionów z genami, typami komórek i rzadkimi komórkami
Z bogatszą referencją w ręku autorzy zapytali, co te regiony robią. Wiele cPeaks leży w pobliżu miejsc startu i zakończenia genów lub pokrywa znane elementy regulacyjne, takie jak promotory, enhancery i miejsca wiązania białek strukturalnych, np. CTCF. Niewielka podgrupa jest dostępna niemal w każdym zbiorze danych; te dłuższe „housekeepingowe” cPeaks zwykle lokują się w regionach promotorów genów potrzebnych do podstawowej konserwacji komórki. Zespół sklasyfikował też cPeaks według tego, jak ostre i spójne są ich granice między próbkami, co odzwierciedla, jak precyzyjnie pobliski DNA jest upakowany w nukleosomy. Regiony o wyraźnie zdefiniowanych brzegach są wzbogacone o konkretne rodziny czynników transkrypcyjnych znanych z przebudowy chromatyny i kierowania rozwojem. Gdy cPeaks użyto jako zestawu cech do analizy wielu zestawów single-cell, poprawiły one dokładność przypisywania typów komórek i były szczególnie pomocne w identyfikacji rzadkich typów komórek oraz subtelnych podtypów, które wcześniejsze zestawy szczytów czy proste siatki genomowe często zacierały.
Śledzenie rozwoju i nowotworów za pomocą wspólnego języka
Moc standardowej referencji staje się oczywista przy porównywaniu bardzo różnych kontekstów biologicznych. Korzystając z cPeaks, autorzy ponownie przeanalizowali dane single-cell z rozwijającej się siatkówki ludzkiej, rozległe atlasy tkanek płodowych i dorosłych oraz kilka nowotworów. Udało się odtworzyć trajektorie rozwojowe i zauważyć, że udział ostro ograniczonych „dobrze pozycjonowanych” cPeaks ma tendencję do wzrostu w etapach przejściowych, a następnie spada, gdy komórki utrwalają stabilne tożsamości. Podobny wzór pojawił się w przebiegu stadium nowotworowego: nowotwory pośrednie wykazywały wyższy udział tych uporządkowanych regionów, sugerując intensywne przebudowywanie regulacyjne. W jednym guzie jajnika cPeaks pomogły ujawnić dwa odrębne subklony komórek nowotworowych z różnymi zmianami liczby kopii DNA, pokazując, jak referencja może odsłonić ukrytą złożoność choroby.
Co to oznacza dla przyszłych badań nad genomem
Dla osób niebędących specjalistami cPeaks można rozumieć jako zunifikowany zestaw współrzędnych wskazujących miejsca, w których genom jest najprawdopodobniej fizycznie otwarty i aktywny w różnych typach ludzkich komórek. Wyrównując nowe eksperymenty pojedynczych komórek chromatyny do tej współdzielonej mapy, badacze mogą porównywać wyniki między studiami, łatwiej dostrzegać rzadkie lub przejściowe stany komórek i zaczynać budować modele regulacji genów w dużej skali — tak jak standaryzowane katalogi genów umożliwiły powstanie atlasów RNA pojedynczych komórek. Obecny katalog cPeaks jest pierwszym szkicem, który będzie się rozrastać wraz z napływem nowych danych, ale już teraz dostarcza wspólnego języka opisu dostępności chromatyny, przybliżając nas do zjednoczonego obrazu, jak upakowanie DNA kieruje rozwojem, zdrowiem i chorobą. 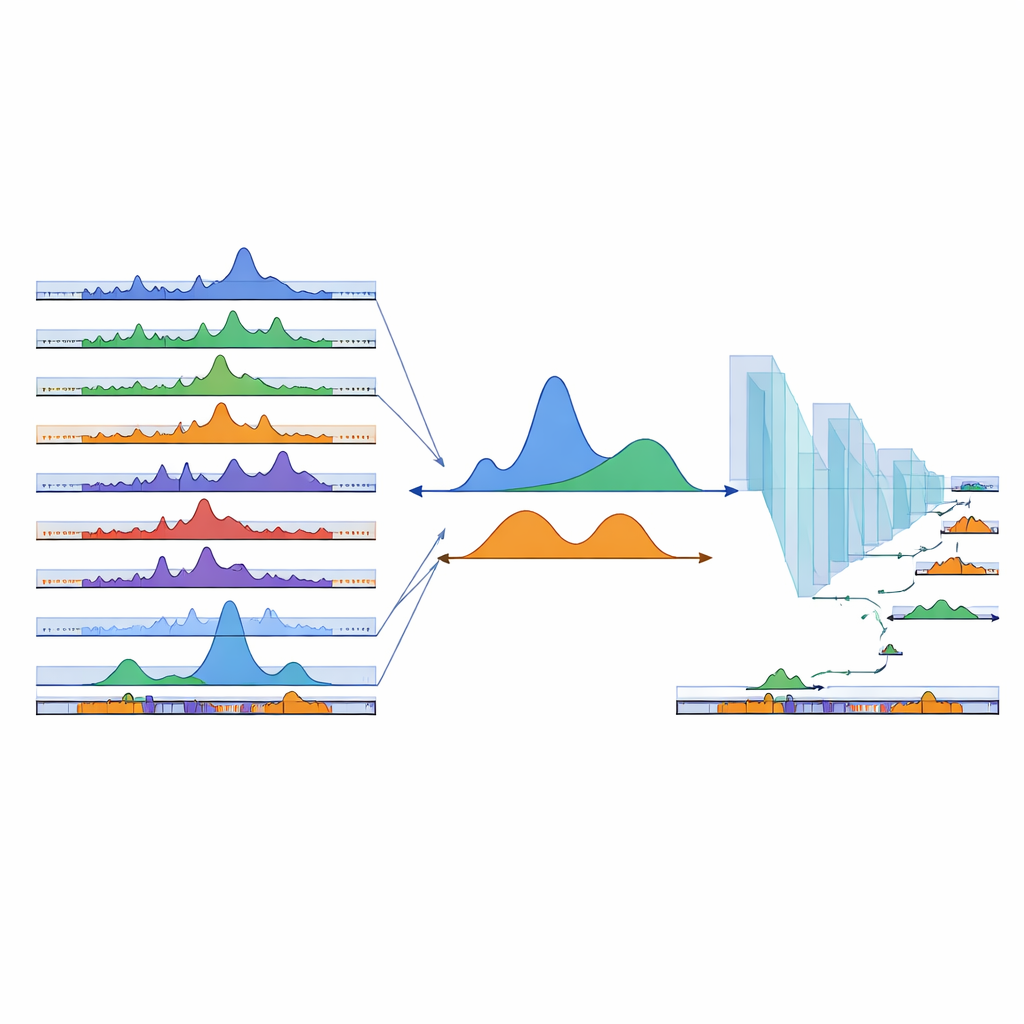
Cytowanie: Meng, Q., Wu, X., Chen, W. et al. A generic reference defined by consensus peaks for single-cell ATAC-seq data analysis. Nat Commun 17, 2522 (2026). https://doi.org/10.1038/s41467-026-69461-6
Słowa kluczowe: dostępność chromatyny, single-cell ATAC-seq, szczyty konsensusu, regulacja genów, głębokie uczenie w genomice