Clear Sky Science · pl
Potrójne celowanie w STING, TGF-β i PD-L1 wzmacnia sygnalizację CXCL16–CXCR6 dla silnej odpowiedzi antynowotworowej
Rozgrzewanie zimnych guzów
Immunoterapia nowotworów zrewolucjonizowała leczenie u niektórych pacjentów, jednak wiele guzów nadal opiera się tym silnym lekom. Badanie to analizuje, dlaczego niektóre nowotwory są oporne na współczesne leki „hamujące punkty kontrolne” i proponuje mądrzejsze, trójskładnikowe działanie, które pobudza obronę organizmu, przyciąga wyspecjalizowane komórki T zabójców i utrzymuje je aktywne bezpośrednio w guzie.
Dlaczego obecne leki immunologiczne nie wystarczają
Większość zatwierdzonych immunoterapii celuje w pojedynczy hamulec komórek odpornościowych, taki jak szlak PD-1/PD-L1. Nowsza klasa leków próbuje pójść dalej, dodatkowo blokując TGF-β, cząsteczkę silnie tłumiącą odporność w zaawansowanych nowotworach. Jeden z takich leków, YM101, łączy blokadę TGF-β i PD-L1 w pojedynczym przeciwciele i dał obiecujące wyniki u myszy. Jednak nawet u genetycznie identycznych zwierząt niektóre guzy ledwie się zmniejszały. Porównując guzy reagujące i oporne, badacze odkryli, że skuteczne leczenie szło w parze z silną aktywacją odporności „wrodzonej”, zwłaszcza sygnalizacją przez szlak zwany STING, który wykrywa nieprawidłowe DNA i uruchamia alarm podobny do antywirusowego.
Dodanie trzeciego dźwigni: szlak STING
Przypuszczając, że słaba aktywacja wrodzona była brakującym elementem, zespół połączył YM101 z doustnym agonistą STING o nazwie MSA-2 w kilku modelach guzów mysich, w tym zwykle trudnych do leczenia „zimnych” guzach. Potrójne podejście — aktywacja STING plus blokada TGF-β i PD-L1 — skuteczniej zmniejszało guzy, wydłużało przeżycie i często chroniło myszy przed odrostem guza po ponownym wyzwaniu, co wskazywało na długotrwałą pamięć immunologiczną. Wyniki przewyższały bardziej konwencjonalne połączenie agonisty STING z blokadą PD-L1 i nawet wzmacniały terapię agonistą STING, gdy blokowano jedynie TGF-β, ujawniając, że sam TGF-β działa jako istotny hamulec dla odporności napędzanej przez STING. 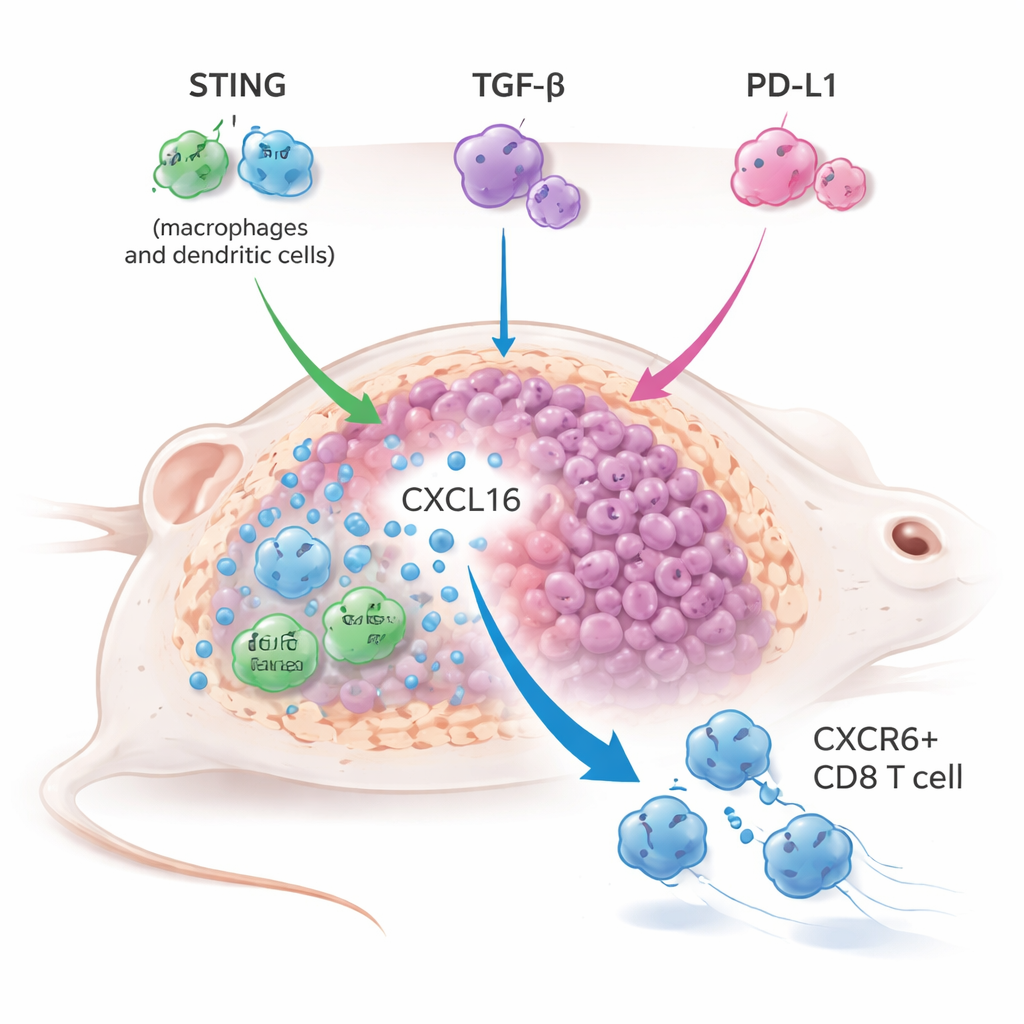
Rekrutowanie wyspecjalizowanej grupy zabójców
Aby zrozumieć, jak działała ta potrójna strategia, badacze zastosowali sekwencjonowanie RNA pojedynczych komórek oraz szczegółowy profil immunologiczny leczonych guzów. Odkryli wyraźną ekspansję określonej podgrupy komórek T zabójczych oznaczonych receptorem CXCR6. Te CXCR6+ CD8 T komórki były silnie uzbrojone, wykazywały wysokie poziomy granzymów, perforyny i cząsteczek zapalnych oraz silne cechy aktywacji i proliferacji. Jednocześnie makrofagi związane z guzem i komórki dendrytyczne zwiększyły produkcję chemokiny CXCL16, która wiąże się z CXCR6 i pomaga zatrzymać te komórki T w guzie. Gdy połączenie CXCL16–CXCR6 zostało przerwane — albo przez blokadę CXCL16, albo przez genetyczne usunięcie CXCR6 w komórkach T — skojarzone leczenie w dużej mierze traciło swoją moc antynowotworową, dowodząc, że ta oś jest kluczowa dla sukcesu terapii.
Jak szlak sygnałowy zostaje włączony
Zagłębiając się dalej, zespół zapytał, jak aktywacja STING i blokada TGF-β wspólnie zwiększają CXCL16. W ludzkich i mysich komórkach odpornościowych agoniści STING silnie zwiększali CXCL16 i przeciwwirusowy cytokin IFN‑β, podczas gdy dodane TGF-β wyraźnie zmniejszało oba. Badacze wykazali, że STING uruchamia sygnalizację IFN‑I, która aktywuje czynnik transkrypcyjny STAT1; STAT1 następnie wiąże się bezpośrednio z regionem kontrolnym genu CXCL16, włączając jego ekspresję. TGF-β zakłóca ten łańcuch, ingerując w kluczowy krok sygnalizacji STING, prawdopodobnie za pośrednictwem białka HDAC4 i reaktywnych form tlenu, tłumiąc aktywację IRF3 oraz późniejszą produkcję IFN‑β i CXCL16. Zablokowanie TGF-β usuwa ten hamulec, co pozwala agonistom STING w pełni zapalić szlak STAT1–CXCL16 w komórkach mieloidalnych i w ten sposób dostarczać sygnały niezbędne, by CXCR6+ komórki T pozostawały i walczyły w guzie.
Budowa jednego precyzyjnego leku
Aby uczynić ten złożony schemat bardziej praktycznym i skoncentrowanym na guzie, badacze zaprojektowali pojedynczy „konjugat przeciwciało‑stymulujący układ immunologiczny” o nazwie Y101S. Ta cząsteczka łączy dwufunkcyjne przeciwciało blokujące TGF-β/PD-L1 z agonistą STING przyłączonym za pomocą rozłączalnego łącznika. Y101S kieruje się do komórek mieloidalnych z ekspresją PD-L1 w guzie, jest internalizowany, a następnie uwalnia lek STING wewnątrz tych komórek. W kilku nowotworach mysich Y101S dorównywał lub przewyższał skuteczność podawania YM101 plus dużej dawki wolnego agonisty STING, mimo że niósł tylko ułamek tej dawki STING. Zwiększał liczbę makrofagów i komórek dendrytycznych CXCL16+, rozszerzał populację CXCR6+ CD8 T, indukował trwałą pamięć immunologiczną i skupiał sygnały zapalne w guzach przy jednoczesnym oszczędzaniu zdrowych narządów, wykazując korzystny profil bezpieczeństwa u myszy. 
Co to oznacza dla przyszłego leczenia nowotworów
Dla niewyspecjalizowanych czytelników kluczowa wiadomość jest taka, że atakowanie nowotworu jednym lub dwoma przełącznikami immunologicznymi może nie wystarczyć — szczególnie gdy guzy czynnie wyciszają wczesne systemy alarmowe. Praca ta pokazuje, że łączenie aktywacji STING z blokadą TGF-β i PD-L1 może przeprogramować mikrośrodowisko guza, mocno przyciągać i utrzymywać wyspecjalizowaną grupę komórek T zabójców oraz osiągać głębsze, trwalsze odpowiedzi w modelach przedklinicznych. Potrójnie celujące przeciwciało‑lek Y101S ucieleśnia tę strategię w pojedynczym, ukierunkowanym preparacie i oferuje mapę drogową dla następnej generacji immunoterapii skierowanych przeciw nowotworom, które obecnie opierają się standardowym lekom hamującym punkty kontrolne.
Cytowanie: Yi, M., Li, T., Gu, Y. et al. Triple targeting of STING, TGF-β, and PD-L1 boosts CXCL16–CXCR6 signaling for potent antitumor response. Nat Commun 17, 1441 (2026). https://doi.org/10.1038/s41467-026-69456-3
Słowa kluczowe: immunoterapia nowotworów, szlak STING, blokada TGF-beta, przeciwciało PD-L1, komórki T CXCL16 CXCR6