Clear Sky Science · pl
Sekwencjonowanie metylacji i hydroksymetylacji DNA w kontekstach chromatynowych występujących jednocześnie
Odczytywanie chemicznych notatek naszych komórek
Każda komórka w organizmie ma tę samą sekwencję DNA, a mimo to komórki mózgowe, skórne i macierzyste zachowują się bardzo różnie. Jednym z powodów jest to, że komórki zapisują na DNA i jego białkowych opakowaniach chemiczne „notatki”, które pomagają włącząć lub wyłączyć geny. Do tej pory naukowcy mieli trudności z odczytaniem kilku takich notatek jednocześnie na tej samej cząsteczce DNA, co pozostawia lukę w naszym rozumieniu ich współdziałania. W tym badaniu przedstawiono nową metodę pozwalającą jednocześnie odczytać zarówno kod genetyczny, jak i kluczowe chemiczne modyfikacje, ujawniając, jak współdziałają, by kontrolować ważne przełączniki DNA zwane wzmacniaczami.
Dlaczego DNA potrzebuje oznaczeń ołówkiem
DNA nie działa w próżni. Jest owinięty wokół białek zwanych histonami, tworząc chromatynę, a zarówno DNA, jak i histony mogą być ozdobione niewielkimi grupami chemicznymi. Dwie ważne modyfikacje na DNA to grupy metylowe i hydroksymetylowe dodane do litery C (cytozyny). Te modyfikacje wpływają na to, jak ciasno DNA jest upakowane i czy pobliskie geny są aktywne. Ogólnie rzecz biorąc, sygnały metylowe często wiążą się z wyciszaniem genów, podczas gdy hydroksymetylacja zwykle występuje tam, gdzie geny są aktywne. Jednak ich wpływ zależy od lokalnego kontekstu: dokładnego miejsca w genomie i sąsiedztwa konkretnych znaków histonowych.
Problem z oddzielnymi mapami
Obecne metody sekwencjonowania potrafią mapować metylację i hydroksymetylację w całym genomie, a inne metody mapują markery histonowe wskazujące na aktywne lub wyciszone obszary. Zwykle wykonuje się je jednak w oddzielnych eksperymentach i porównuje komputerowo. To informuje, które cechy mają tendencję do współwystępowania w tej samej okolicy, ale nie odpowiada na pytanie, czy naprawdę współistnieją na tej samej cząsteczce DNA w pojedynczej komórce. Starsze próby łączenia tych pomiarów opierały się na silnych zabiegach chemicznych, które uszkadzały DNA i, co kluczowe, nie pozwalały niezawodnie rozróżnić metylacji od hydroksymetylacji na tym samym odczycie. W rezultacie badaczom brakowało jasnego, molekularnego obrazu tego, jak kombinacje znaków współpracują.
Nowa metoda odczytu wielowarstwowego
Autorzy opracowali metodę nazwaną 6-base-CUT&Tag, która potrafi odczytać wszystkie cztery litery DNA oraz dwa stany chemiczne cytozyny — zwykłą, metylowaną i hydroksymetylowaną — na fragmentach DNA fizycznie związanych z wybranymi cechami chromatyny. Najpierw wykorzystują przeciwciała jak molekularne haczyki, aby wyłowić DNA owinięte wokół histonów niosących konkretny znak, na przykład znak chromatyny aktywnej. Zmodyfikowany enzym następnie wstawia specjalne adaptery, przekształcając każdy wychwycony fragment DNA w małą pętlę, która przetrwa etapy oczyszczania niszczące luźne kawałki. Udoskonalony proces chemiczny i enzymatyczny konwertuje różne stany cytozyny na rozróżnialne sygnały sekwencyjne, które nowoczesne sekwencjonery potrafią odczytać. W ten sposób pojedynczy odczyt informuje, skąd pochodził fragment, jaki znak histonowy niósł i które cytozyny były zmetylowane lub zhydroksymetylowane.
Zbliżenie na przełączniki genów
Używając mysich embrionalnych komórek macierzystych jako przypadku testowego, zespół zastosował 6-base-CUT&Tag do kilku kluczowych znaków histonowych oznaczających różne rodzaje regulatorowego DNA. Skoncentrowali się na wzmacniaczach — odcinkach DNA działających jak przełączniki kontrolujące, kiedy i gdzie geny się włączają. Wzmacniacze mogą występować w stanie „aktywnym”, „przygotowanym” (primed) lub „zaprężonym” (poised), rozróżnianym przez konkretne znaki histonowe. Badacze odkryli, że wzmacniacze oznaczone wyłącznie przez znak histonowy H3K4me1 (często uważany za „przygotowany”) miały najwyższe poziomy zarówno metylacji, jak i hydroksymetylacji DNA, zwłaszcza gdy badano je bezpośrednio na nukleosomach związanych z H3K4me1. W przeciwieństwie do tego, wzmacniacze z dodatkowymi oznakami silnej aktywności lub represji miały mniej tych modyfikacji DNA lub wykazywały przesunięcie w kierunku hydroksymetylacji, co sugeruje trwające usuwanie znaków metylowych.
Odczytywanie stanów wzmacniaczy z większą precyzją
Ponieważ wszystkie typy wzmacniaczy dzielą znak H3K4me1, zespół zapytał, czy szczegółowy wzór modyfikacji DNA specyficzny dla DNA oznaczonego H3K4me1 może samodzielnie rozróżnić różne stany wzmacniaczy. Wytrenowali model uczenia maszynowego używając danych z 6-base-CUT&Tag, aby klasyfikować wzmacniacze jako aktywne, przygotowane lub zaprężone, wyłącznie na podstawie ilości metylu i hydroksymetylu obecnych przy tym pojedynczym znaku histonowym. Model ten przewyższył inny, w przeciwnym razie identyczny model wytrenowany na standardowych danych z całego genomu, które nie są ograniczone do żadnego znaku histonowego. Innymi słowy, odczytywanie modyfikacji DNA w bezpośrednim kontekście ich występowania daje ostrzejszy obraz niż uśrednianie danych z całego DNA w komórce.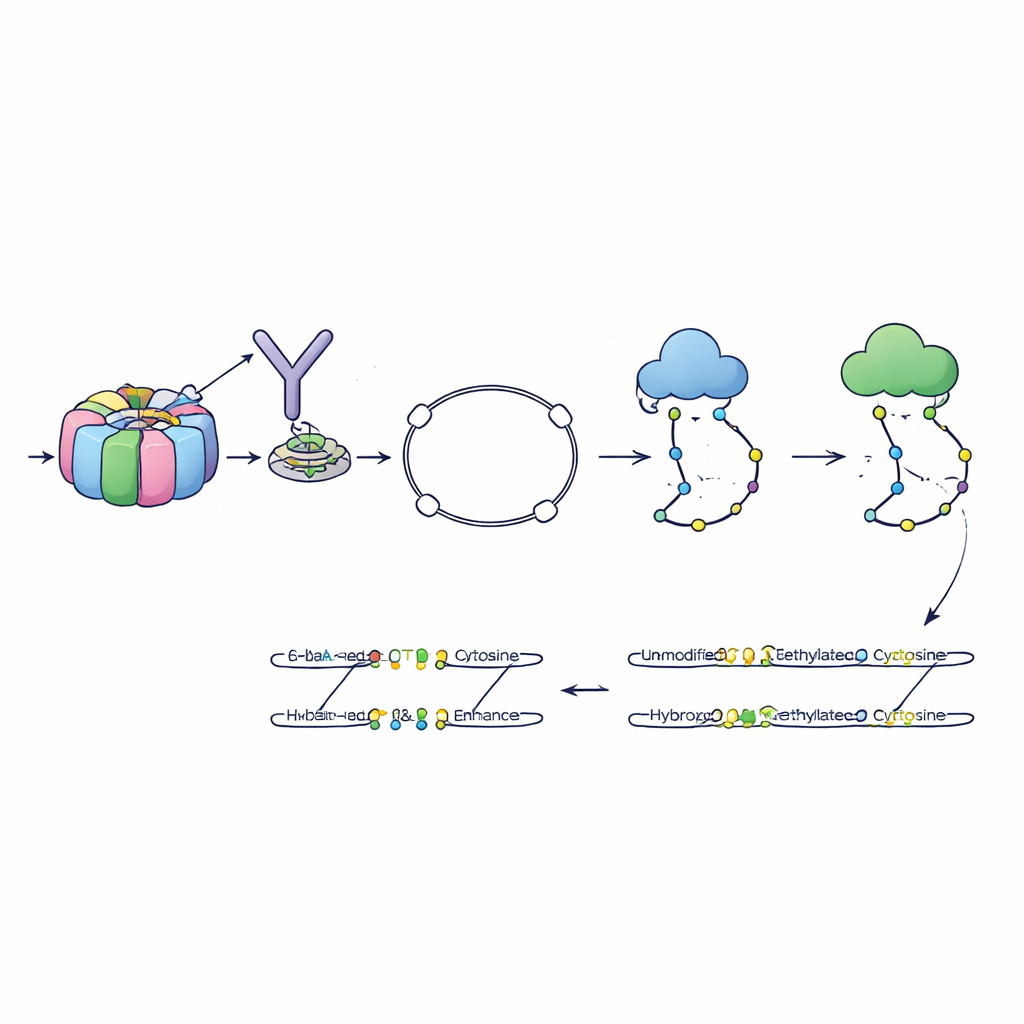
Co to oznacza dla zrozumienia tożsamości komórek
Dla czytelnika niebędącego specjalistą kluczowy wniosek jest taki, że ta metoda pozwala naukowcom odczytać kilka warstw informacji — sekwencję DNA, modyfikacje DNA i znaki histonowe — na tej samej cząsteczce. Ten szczegółowy obraz ujawnia, jak konkretne kombinacje chemicznych znaczników definiują gotowość przełączników genowych w komórkach macierzystych. Ponieważ 6-base-CUT&Tag jest bardziej wydajna i mniej niszcząca niż starsze podejścia, może odkrywać subtelne wzorce, które wcześniej były ukryte. Z czasem to wielowarstwowe czytanie chromatyny może pomóc wyjaśnić, jak komórki zapamiętują swoją tożsamość, jak zmieniają się w czasie rozwoju lub choroby oraz jak można precyzyjniej celować w kod regulacyjny w terapii.
Cytowanie: Araujo Tavares, R.d.C., Dhir, S., He, X. et al. Sequencing DNA methylation and hydroxymethylation at co-occurring chromatin features. Nat Commun 17, 2591 (2026). https://doi.org/10.1038/s41467-026-69429-6
Słowa kluczowe: epigenetyka, metylacja DNA, chromatyna, wzmacniacze, komórki macierzyste