Clear Sky Science · pl
CD38 degraduje MAVS przez mitofagię, aby zahamować wydzielanie interferonu typu I w komórkach raka nosogardzieli i osłabia odporność przeciwnowotworową zależną od limfocytów CD8+
Dlaczego to ma znaczenie dla leczenia raka
Rak nosogardzieli rozwija się za nosem i jest szczególnie częsty w Azji Wschodniej i Południowo-Wschodniej. Leki immunologiczne uwalniające własne limfocyty T organizmu zmieniły rokowanie u niektórych pacjentów, lecz większość wciąż nie odnosi korzyści. Badanie ujawnia ukrytą blokadę wewnątrz samych komórek nowotworowych: cząsteczkę nazwaną CD38, która dyskretnie wyłącza wewnętrzny system alarmowy i osłabia atak zabójczych limfocytów CD8. Zrozumienie i unieszkodliwienie tej blokady może sprawić, że istniejące immunoterapie będą skuteczne u znacznie większej liczby pacjentów.
Ukryty przełącznik na komórkach nowotworowych
Naukowcy skupili się na CD38, białku występującym na wielu komórkach układu odpornościowego, ale także obecnym na komórkach raka nosogardzieli. Wcześniejsze badania powiązały CD38 z opornością na popularne leki blokujące punkty kontrolne, które celują w PD-1 i PD-L1. Zespół sprawdził, czy CD38 obecne wewnątrz komórek nowotworowych bezpośrednio zmienia zdolność limfocytów CD8 do rozpoznania i zniszczenia tych komórek. Hodując ludzkie komórki nowotworowe z CD38 lub bez niego razem z aktywowanymi ludzkimi limfocytami CD8, stwierdzili, że usunięcie CD38 z komórek nowotworowych znacząco zwiększało moc T komórek: wydzielały one więcej kluczowych cząsteczek ataku, lepiej przeżywały i zabijały więcej komórek nowotworowych. Przywrócenie CD38 powodowało spadek funkcji T, wskazując na CD38 jako wewnętrzny hamulec ataku immunologicznego.
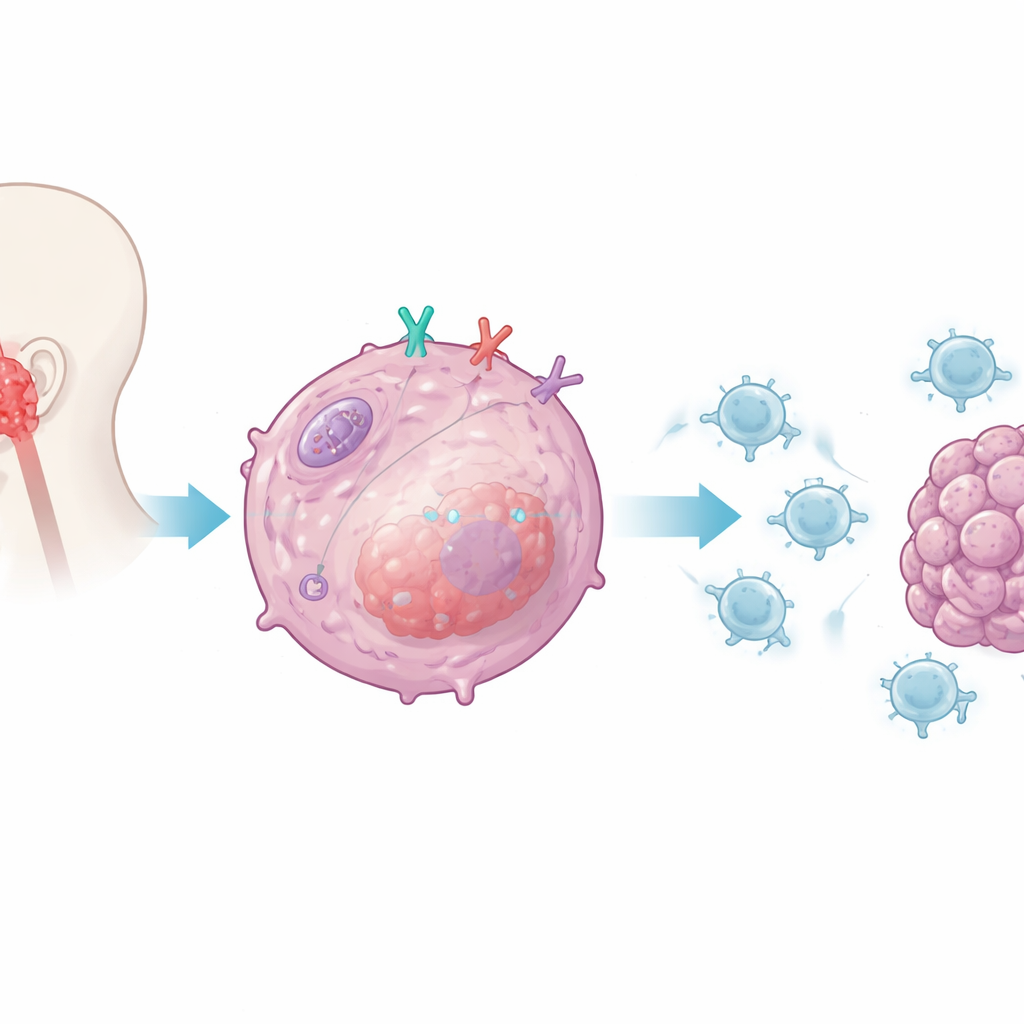
Przyciszanie wewnętrznego alarmu komórkowego
Zespół następnie zbadał, jak CD38 przekazuje ten sygnał tłumiący. Skoncentrowali się na wrodzonym systemie alarmowym komórki, który zwykle wykrywa materiał genetyczny przypominający wirus i uruchamia interferony typu I — silne przekaźniki stymulujące układ odpornościowy. W komórkach nowotworowych pozbawionych CD38 zaobserwowano wyraźny wzrost interferonu beta oraz chemokin przyciągających limfocyty CD8 do guza. Wykazali, że CD38 selektywnie tłumi szlak kontrolowany przez wewnętrzny sensor RIG-I i jego białko adaptorowe MAVS, które znajduje się na mitochondriach — fabrykach energetycznych komórki. Gdy CD38 było obecne, aktywacja tego szlaku i jego molekularnych efektorów była stłumiona; po usunięciu CD38 sygnalizacja i produkcja interferonu nasilały się, zwiększając widoczność guza dla układu odpornościowego.
Jak CD38 niszczy kluczowy węzeł sygnałowy
Wnikliwsze badania wykazały, że CD38 fizycznie wiąże się z MAVS na mitochondriach i zakłóca partnerstwo MAVS z RIG-I, osłabiając przekazywanie sygnału. Co istotniejsze, wyższe poziomy CD38 prowadziły do zmniejszenia ilości białka MAVS bez zmiany jego sekwencji genetycznej, co sugeruje aktywną degradację. Testy z różnymi inhibitorami pokazały, że ten ubytek zależy od mechanizmów recyklingu komórkowego znanych jako autofagia, a konkretnie od formy ukierunkowanej na mitochondria. CD38 zwiększało markery „autofagii mitochondrialnej”, zmniejszało poziomy kilku białek mitochondrialnych i promowało pakowanie MAVS do struktur autofagosomów, które następnie są rozkładane. Blokada mitochondrialnej autofagii zachowywała MAVS i przywracała sygnalizację interferonową, co wskazuje, że CD38 wyłącza alarm przez kierowanie MAVS do szlaku degradacji komórkowej.
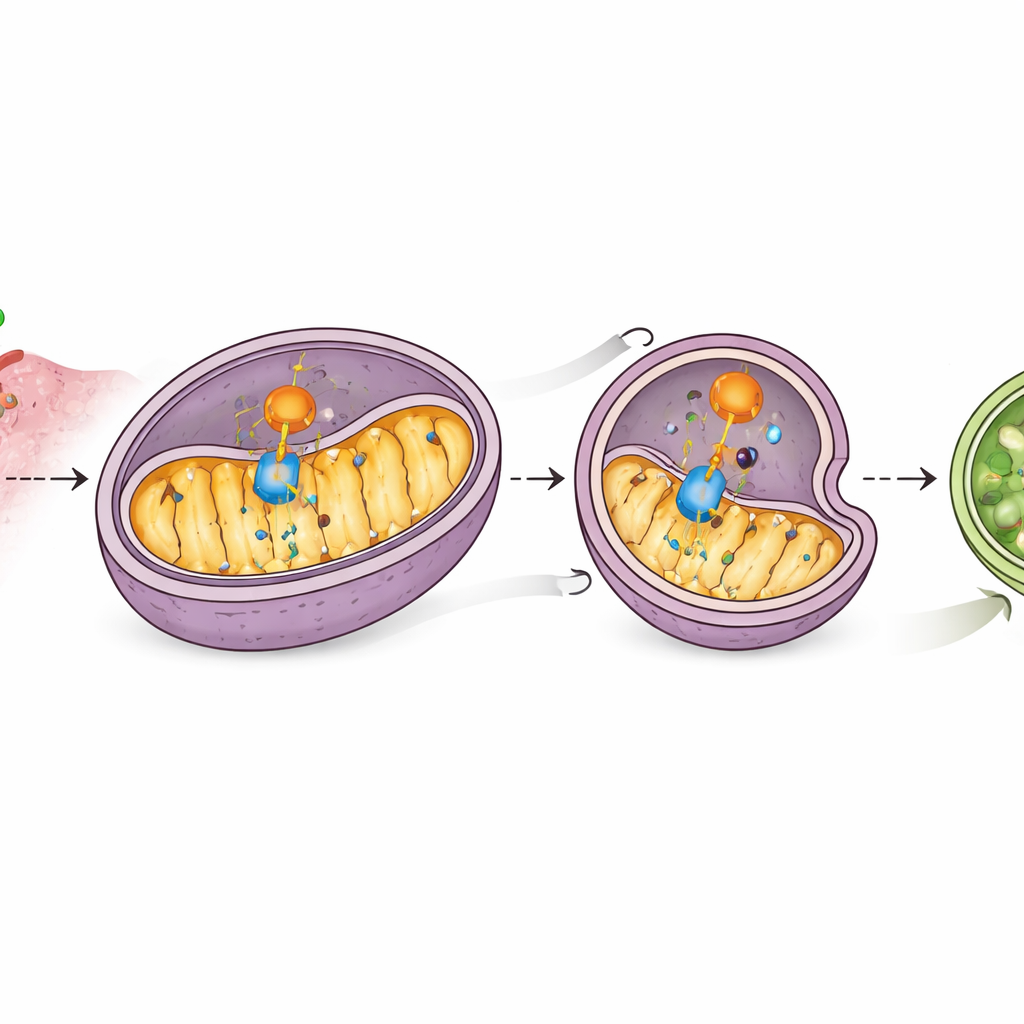
Pomocnik kierujący mitochondria do samozniszczenia
Badanie zidentyfikowało kolejnego uczestnika — PHB2, białko mitochondrialne działające jako receptor dla ukierunkowanej usuwalności mitochondriów. Spektrometria mas i eksperymenty wiążące wykazały, że CD38 wchodzi w interakcję z PHB2 i zwiększa obecność PHB2 w mitochondriach, gdzie PHB2 z kolei rekrutuje podstawowe elementy maszynerii autofagii. PHB2 również wiąże MAVS, a CD38 wzmacnia to łącze. Gdy poziom PHB2 został zmniejszony, CD38 nie mogło już skutecznie wywołać autofagii mitochondrialnej, poziomy MAVS odrosły, a geny związane z interferonem zostały ponownie aktywowane. To ujawnia łańcuch zdarzeń: CD38 angażuje PHB2, PHB2 angażuje MAVS, a wspólnie kierują MAVS do mitochondriów przeznaczonych do degradacji, uciszając alarm interferonowy.
Dowody z modeli zwierzęcych
Aby sprawdzić wpływ w organizmach żywych, badacze użyli mysich guzów zaprojektowanych tak, by nie zawierały CD38. U myszy z prawidłowym układem odpornościowym takie guzy rosły wolniej, zawierały więcej limfocytów CD8 i miały wyższy odsetek komórek produkujących interferon-gamma, co jest znamieniem aktywnej odpowiedzi przeciwnowotworowej. Zablokowanie receptora dla interferonów typu I usuwało tę przewagę, potwierdzając, że sygnalizacja interferonowa jest niezbędna dla wzmocnionej odporności. Umys o humanizowanym układzie odpornościowym z guzami nosogardzieli zmniejszenie CD38 podobnie spowolniło wzrost i zwiększyło infiltrację limfocytów CD8, lecz korzyść ta znikała, gdy w komórkach guza również obniżono MAVS. Wyniki in vivo utrwalają koncepcję, że oś CD38–PHB2–MAVS w komórkach nowotworowych kształtuje siłę odpowiedzi limfocytów T gospodarza.
Co to oznacza dla przyszłych terapii
Podsumowując, praca pokazuje, że CD38 w komórkach raka nosogardzieli działa jak wewnętrzny sabotażysta odporności przeciwnowotworowej. Poprzez uruchomienie selektywnej formy recyklingu mitochondrialnego CD38 zmniejsza ilość MAVS, osłabia produkcję interferonu typu I, obniża prezentację antygenów i ostatecznie tłumi atak limfocytów CD8. Obecne związki blokujące CD38 głównie celują w jego aktywność enzymatyczną i nie usuwają białka ani nie przywracają MAVS. Autorzy postulują, że nowe strategie mające na celu obniżenie poziomu CD38 lub przerwanie jego partnerstwa z PHB2 lub MAVS mogłyby ponownie wzbudzić alarm interferonowy wewnątrz guzów. W połączeniu z istniejącymi inhibitorami punktów kontrolnych takie podejścia mogą przekształcić więcej nowotworów nosogardzieli — a potencjalnie także innych — z „zimnych” immunologicznie w odpowiednie na immunoterapię.
Cytowanie: Liang, L., Li, W., Liu, S. et al. CD38 degrades MAVS through mitophagy to inhibit type I interferon secretion in nasopharyngeal carcinoma cells and impairs CD8+T cell-mediated anti-tumor immunity. Nat Commun 17, 2544 (2026). https://doi.org/10.1038/s41467-026-69339-7
Słowa kluczowe: rak nosogardzieli, immunoterapia nowotworów, interferon typu I, limfocyty CD8, mitofagia