Clear Sky Science · pl
Warianty powodujące utratę funkcji aktywatora CAPN1, CD99L2, wywołują sprzężoną z chromosomem X spastyczną ataksję
Dlaczego ma to znaczenie dla rodzin z niewyjaśnionymi problemami ruchowymi
Wiele osób przez lata żyje z niewyjaśnionymi trudnościami w chodzeniu, sztywnością mięśni albo zaburzeniami równowagi i mowy, nie poznając prawdziwej przyczyny. To badanie pokazuje, jak nowoczesne testy DNA mogą wreszcie dać odpowiedzi niektórym z tych rodzin. Naukowcy nie tylko porównali różne badania genetyczne stosowane w rzadkich zaburzeniach ruchu, lecz także odkryli wcześniej nieznaną przyczynę schorzenia zwanego sprzężoną z chromosomem X spastyczną ataksją, wskazując na szlaki biologiczne, które mogą mieć znaczenie także w bardziej powszechnych chorobach mózgu.
Znajdowanie genetycznych igieł w stogu siana rzadkich chorób
Rzadkie zaburzenia ruchu, takie jak ataksja (niepewne ruchy) i spastyczna paraplegia (sztywne, słabe nogi), często podejrzewa się o pochodzenie genetyczne, ale u większości pacjentów standardowe testy nie wykrywają przyczyny. Zespół śledził 2 811 osób w Niemczech i w całej Europie, które przez sześć lat były kierowane z podejrzeniem rzadkiego zaburzenia ruchu. Najpierw zastosowano tradycyjne testy ukierunkowane, które poszukują znanych ekspansji powtórzeń w kilku genach; dały one odpowiedź w około 11% przypadków. Następnie użyto sekwencjonowania eksomowego, które odczytuje jedynie białkodajne części genomu, i znaleziono jednoznaczne wyjaśnienia genetyczne u około 19% pacjentów, zwłaszcza u tych ze spastycznością.
Wyjście poza standardowe testy dzięki sekwencjonowaniu całego genomu
Aby pójść dalej, naukowcy zastosowali sekwencjonowanie całego genomu, które odczytuje niemal cały materiał DNA danej osoby, w tym regiony, które mogą umykać standardowym testom i eksomom. W grupie 486 osób, które przeszły to bardziej wszechstronne badanie, odsetek rozpoznań wzrósł o około 7,5 punktu procentowego, głównie dlatego, że sekwencjonowanie genomu lepiej wykrywa złożone zmiany, takie jak przemieszczenia strukturalne i ekspansje powtórzeń. Badanie wykazało również, że starannie zebrane informacje kliniczne — zwłaszcza konkretne opisy objawów, młodszy wiek w momencie badania oraz występowanie spastyczności w połączeniu z innymi problemami ruchowymi — pomagały przewidzieć, kto najprawdopodobniej otrzyma jednoznaczną genetyczną diagnozę.
Odkrycie nowej sprzężonej z chromosomem X przyczyny spastycznej ataksji
Nawet po tych szerokich badaniach wielu pacjentów pozostało bez rozpoznania. Naukowcy połączyli dane genetyczne z ponad 13 000 osób i zastosowali podejście „obciążenia genowego”, pytając, które geny noszą podejrzane warianty częściej u pacjentów niż u zdrowych kontrolnych. Ta analiza wskazała nie tylko znane geny chorobowe, ale także silnie wyeksponowała wcześniej pomijany gen na chromosomie X o nazwie CD99L2. Łącząc wyniki z kilku rodzin z Europy, zidentyfikowali 25 dotkniętych mężczyzn z 20 rodzin, którzy mieli uszkadzające warianty w tym genie. Ci mężczyźni zwykle rozwijali problemy z chodzeniem, sztywność nóg, niewyraźną mowę i czasami trudności z równowagą w średnim lub późniejszym wieku dorosłym, podczas gdy nosicielki płci żeńskiej były na ogół bezobjawowe — wzorce zgodne z chorobą sprzężoną z X. Warianty przede wszystkim niszczyły normalne białko lub usuwały kluczowe jego części, co mocno sugeruje, że utrata funkcji tego białka wywołuje chorobę.
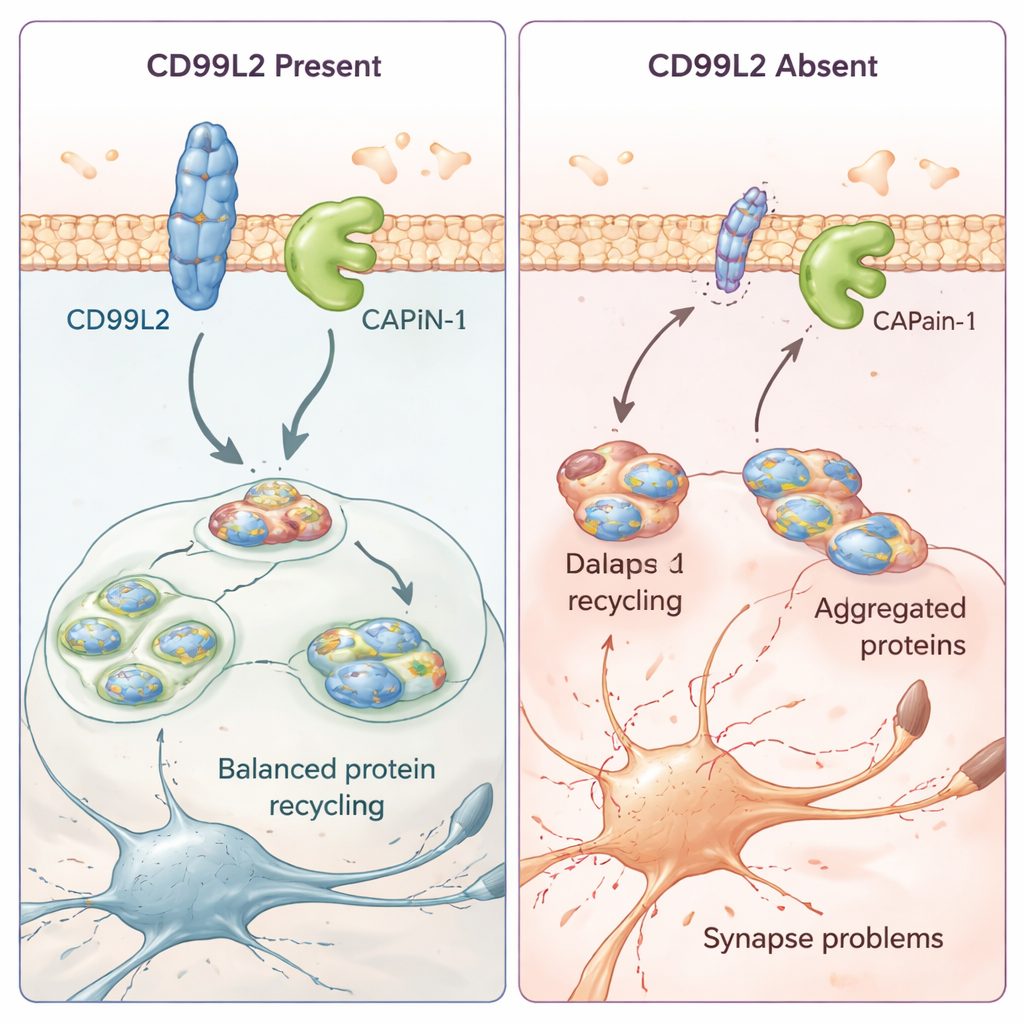
Jak małe białko błonowe pomaga chronić komórki mózgu
Aby zrozumieć, co CD99L2 robi w komórkach, zespół sięgnął po modele komórkowe i komórki skórne pochodzące od pacjentów. Odkryli, że białko CD99L2 znajduje się w błonie komórkowej i zwykle jest znakowane małymi „ubikwitynowymi” znaczkami, które kontrolują, jak długo przetrwa zanim zostanie rozłożone. CD99L2 fizycznie wiąże się z kalpainą-1 (CAPN1), enzymem aktywowanym przez wapń, który przycina inne białka i pomaga utrzymać synapsy — punkty kontaktowe między komórkami nerwowymi — w dobrym stanie. Gdy CD99L2 jest obecny i nienaruszony, pomaga w kontrolowany sposób włączać i wyłączać kalpainę-1, a następnie sam jest obcinany i poddawany recyklingowi. Gdy CD99L2 jest nieobecny lub strukturalnie zmieniony, aktywacja kalpainy-1 jest zaburzona. W komórkach pacjentów idzie to w parze z zakłóconą aktywnością wielu genów związanych z synapsami i komunikacją neuronów, co sugeruje, że subtelne, ale szeroko zakrojone zmiany w obwodach mózgowych mogą leżeć u podstaw stopniowo postępujących problemów ruchowych.
Co to oznacza dla pacjentów dziś i w przyszłości
Dla rodzin z niewyjaśnioną spastyczną ataksją lub spastyczną paraplegią ta praca przynosi dwa rodzaje postępu. Po pierwsze, pokazuje, że wczesne zastosowanie sekwencjonowania całego genomu, w połączeniu ze starannym opisem klinicznym, może wyraźnie zwiększyć szanse na pewne genetyczne rozpoznanie. Po drugie, dodaje CD99L2 do listy genów kontrolujących aktywność kalpainy, szlaku już powiązanego z innymi rzadkimi ataksjami oraz z powszechnymi schorzeniami, takimi jak choroba Alzheimera i Parkinsona. W praktycznym ujęciu badanie ujawnia nowy „włącznik–wyłącznik”, który pomaga utrzymać równowagę mechanizmów naprawczych komórek mózgowych; gdy ten przełącznik zostaje uszkodzony, neurony stopniowo ulegają pogorszeniu, prowadząc do sztywności i zaburzeń koordynacji. Zrozumienie tego przełącznika może w końcu otworzyć drogę do terapii, które precyzyjnie dostrajają aktywność kalpainy i chronią komórki mózgowe w szeregu chorób neurologicznych.
Cytowanie: Menden, B., Incebacak Eltemur, R.D., Demidov, G. et al. Loss-of-function variants in the CAPN1 activator CD99L2 cause X-linked spastic ataxia. Nat Commun 17, 1698 (2026). https://doi.org/10.1038/s41467-026-69337-9
Słowa kluczowe: spastyczna ataksja, rzadkie zaburzenia ruchu, sekwencjonowanie genomu, CD99L2, kalpaina-1