Clear Sky Science · pl
Celowanie w NHEJ aktywuje sygnalizację STING poprzez degradację MYC, by wzmocnić odpowiedź przeciwnowotworową w SCLC
Dlaczego te badania są ważne
Rak drobnokomórkowy płuca jest jednym z najbardziej śmiertelnych nowotworów — u większości chorych przeżycie wynosi mniej niż rok od rozpoznania. Co zaskakujące, guzy te mają wiele mutacji DNA, które teoretycznie powinny czynić je łatwymi celami dla układu odpornościowego, jednak w praktyce słabo reagują na współczesne leki immunoterapeutyczne. W pracy tej odkryto ukrytą, molekularną blokadę, która uniemożliwia układowi odpornościowemu rozpoznanie tych guzów, oraz pokazano, że wyłączenie kluczowego białka naprawczego DNA może przełączyć te nowotwory z immunologicznie „zimnych” na „gorące”, co pozwala na znacznie lepsze działanie istniejących terapii.
Ukryty przełącznik naprawczy w guzach płuca
Naukowcy rozpoczęli od przeszukania danych genetycznych z ponad 179 000 ludzkich guzów obejmujących 24 typy nowotworów. Skupili się na szlaku naprawy DNA zwanym niehomologicznym łączeniem końców (NHEJ), który naprawia groźne pęknięcia nici DNA. Centralnym regulatorem tego szlaku okazało się białko DNAPKcs (kodowane przez gen PRKDC), którego poziom był wyjątkowo wysoki w raku drobnokomórkowym płuca. Wśród tysięcy próbek guzów płuca przypadki drobnokomórkowe wykazywały najsilniejszą aktywność tego „przełącznika” naprawczego. Pacjenci, u których guzy miały najwyższe poziomy PRKDC, żyli krócej i rzadziej odnosili korzyść z chemioterapii oraz inhibitorów punktów kontrolnych immunologicznych, co sugeruje, że DNAPKcs pomaga guzom przetrwać zarówno uszkodzenia DNA, jak i ataki immunologiczne.
Od uszkodzenia DNA do wewnętrznego alarmu
Aby sprawdzić, co się dzieje po wyłączeniu tego przełącznika, zespół zastosował zarówno leki, jak i narzędzia wyciszające geny, aby zablokować DNAPKcs w panelach komórek raka drobnokomórkowego oraz w modelach guza u myszy. W wielu z tych modeli, szczególnie w tych przypominających ludzkie podtypy o wysokiej aktywności onkogenu MYC, inhibitory DNAPKcs znacząco ograniczały wzrost komórek nowotworowych, a nawet zmniejszały guzy pochodzące od pacjentów u myszy. Na poziomie komórkowym blokada DNAPKcs prowadziła do nagromadzenia uszkodzeń DNA, widocznych jako punkty markera uszkodzeń w jądrze oraz jako drobne dodatkowe ciała wypełnione DNA, tzw. mikronuklei. Fragmenty te przedostawały się do cytoplazmy, gdzie mogły być rozpoznawane jako sygnały niebezpieczeństwa.
Włączenie komórkowego „wirusowego” systemu alarmowego
Wolne DNA w niewłaściwym miejscu zwykle świadczy o zakażeniu wirusowym. Komórki wykrywają je za pomocą sensora zwanego cGAS, który uruchamia dalszy szlak alarmowy nazwany STING. Autorzy pokazali, że po zahamowaniu DNAPKcs cGAS gromadził się na mikronukleach, STING ulegał aktywacji, a włączona została kaskada cząsteczek pobudzających układ odpornościowy. Komórki zwiększyły produkcję interferonów typu I i II oraz chemokin przyciągających komórki odpornościowe. Zwiększyła się też ekspresja na powierzchni kluczowych „flag” (cząsteczek MHC klasy I), które pomagają komórkom odpornościowym rozpoznawać antygeny nowotworowe. Gdy szlak STING był chemicznie zablokowany lub genetycznie wyciszony, te zmiany prawie znikały, a przeciwnowotworowe efekty hamowania DNAPKcs były znacznie osłabione, co podkreśla, że ten wewnętrzny system alarmowy jest niezbędny dla odpowiedzi.
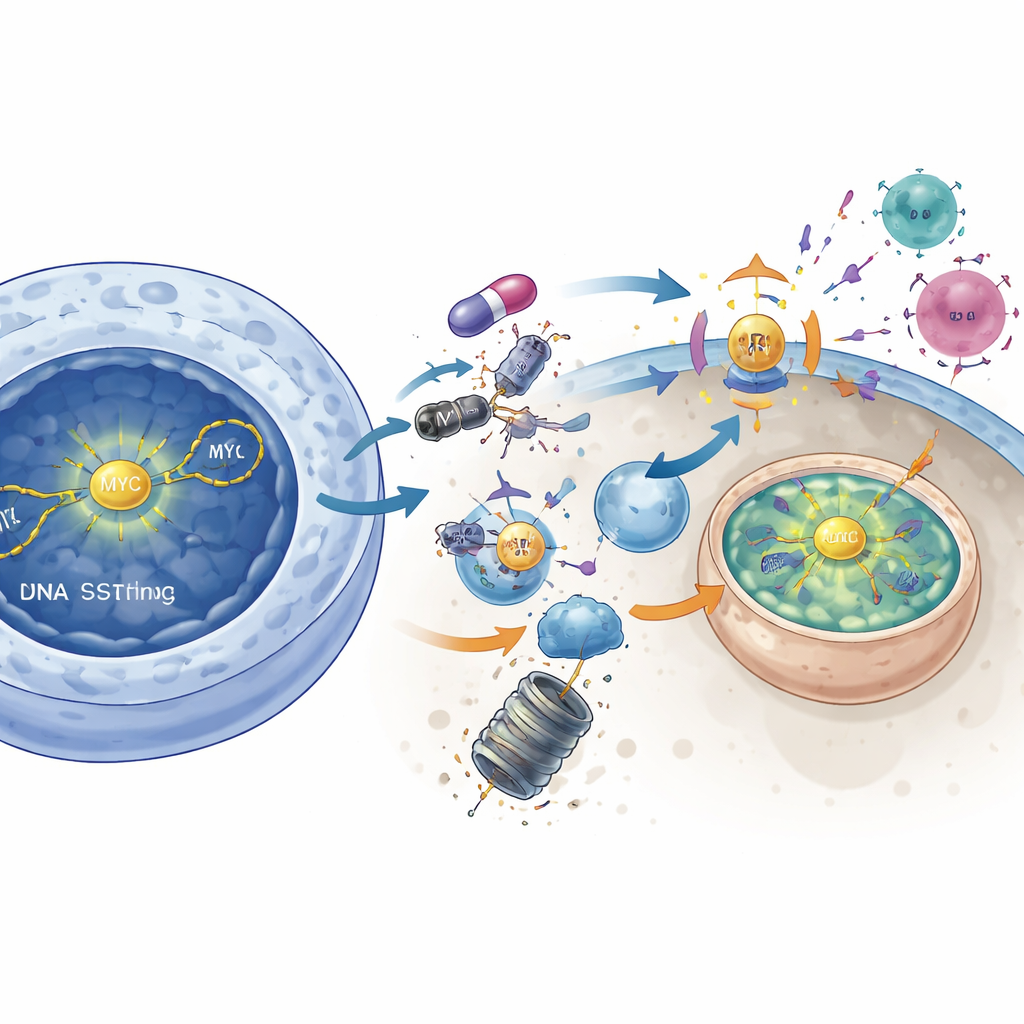
Unieszkodliwienie MYC, by obnażyć guz
Badanie dodatkowo łączy DNAPKcs z silnym czynnikiem wzrostu MYC, białkiem uznawanym długo za „nieuleczalne”. W guzach o wysokiej aktywności MYC hamowanie DNAPKcs zmniejszało aktywne sygnalizowanie AKT i uwalniało molekularną blokadę innego enzymu, GSK3β. Po aktywacji GSK3β oznaczało MYC do zniszczenia, co prowadziło do spadku poziomów białka MYC. Bezpośrednie obniżenie MYC za pomocą narzędzi genetycznych naśladowało wiele immunostymulujących efektów blokady DNAPKcs: wzrastała sygnalizacja STING, włączone były geny interferonowe, a MHC klasy I rosło. Odwrotnie, wymuszone nadmierne wytwarzanie MYC w komórkach w dużej mierze niwelowało immunoumacniający wpływ inhibitora DNAPKcs. To sugeruje, że DNAPKcs normalnie pomaga stabilizować MYC, a skierowanie MYC na degradację jest kluczowym krokiem w uruchamianiu odpowiedzi przeciwnowotworowej.
Od „zimnych” do „gorących” guzów w modelach in vivo
W immunokompetentnych modelach mysich, które wiernie odzwierciedlają ludzki rak drobnokomórkowy płuca, leczenie samym inhibitorem DNAPKcs znacząco spowalniało lub zmniejszało guzy. Co ważne, połączenie inhibitora z istniejącym lekiem przeciw PD-L1 przekształciło wcześniej oporne guzy, prowadząc do dramatycznej regresji guza, a w niektórych przypadkach do całkowitego zaniku. Szczegółowy profil immunologiczny wykazał, że hamowanie DNAPKcs zwiększało liczbę komórek CD8 zabijających nowotwór, wzmacniało prozapalne makrofagi M1, redukowało komórki tłumiące oraz podnosiło poziomy MHC klasy I w guzach. Usunięcie limfocytów CD8 lub wyłączenie STING odwracało te korzyści, potwierdzając, że terapia działa przez przekształcenie guza w punkt przyciągający atak immunologiczny, a nie jedynie przez bezpośrednie zabijanie komórek nowotworowych.
Co to oznacza dla pacjentów
W sumie wyniki te wskazują na DNAPKcs jako centralnego koordynatora zarówno naprawy DNA, jak i unikania nadzoru immunologicznego w raku drobnokomórkowym płuca. Blokując DNAPKcs, guzy gromadzą uszkodzenia DNA, MYC zostaje destabilizowany, alarm cGAS–STING jest uruchamiany, a szlaki interferonowe i prezentacji antygenu włączane. Ten ciąg zdarzeń przekształca immunologicznie ciche guzy w takie, które silnie reagują na inhibitory punktów kontrolnych i chemioterapię w modelach przedklinicznych. Choć potrzebne są jeszcze badania kliniczne, praca ta sugeruje, że istniejące inhibitory DNAPKcs mogłyby być łączone z immunoterapią, dając pacjentom z tym agresywnym nowotworem lepszą szansę na długotrwałą kontrolę choroby.
Cytowanie: Chakraborty, S., Elliott, A., Sen, U. et al. Targeting NHEJ activates STING signaling through MYC degradation to boost antitumor immunity in SCLC. Nat Commun 17, 2597 (2026). https://doi.org/10.1038/s41467-026-69262-x
Słowa kluczowe: rak drobnokomórkowy płuca, hamowanie naprawy DNA, szlak STING, degradacja MYC, immunoterapia nowotworów