Clear Sky Science · pl
scLong: model bazowy o miliardzie parametrów do uchwycenia kontekstu długozasięgowego genów w transkryptomice pojedynczych komórek
Nauczanie komputerów odczytywania ukrytego języka komórek
Każda komórka w twoim ciele jest jak tętniące życiem miasto, w którym geny włączają się i wyłączają w złożonych wzorcach. Nowoczesne sekwencjonowanie RNA pojedynczych komórek potrafi teraz nasłuchiwać aktywności każdej komórki z osobna, ale wynikiem jest przytłaczający potok liczb. W artykule przedstawiono scLong, rozbudowany model sztucznej inteligencji zaprojektowany do interpretacji tych skomplikowanych wzorców aktywności genów, łącznie z subtelnymi sygnałami, które starsze metody zwykle pomijają. Jego celem jest pomoc badaczom w zrozumieniu, jak komórki reagują na wyciszenie genów, dodanie leków lub rozwój choroby.
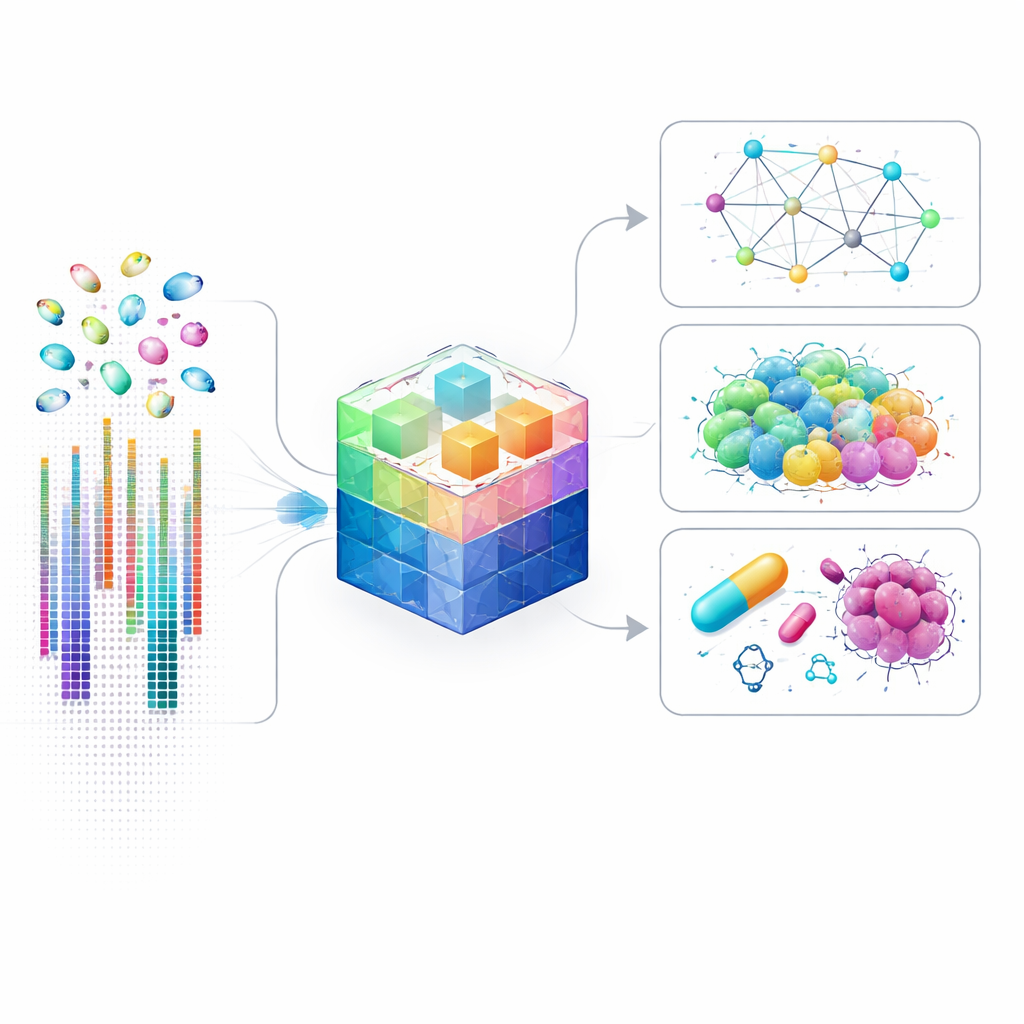
Dlaczego mapy genów na poziomie komórki są ważne
Tradycyjne badania genów często łączą miliony komórek, uśredniając wyniki i zacierając rzadkie lub nietypowe komórki. Techniki pojedynczych komórek zmieniły to podejście, mierząc aktywność genów w każdej komórce osobno: ujawniły ukryte typy komórek, subtelną komunikację między nimi oraz szczegółowe obwody sterujące decydujące o losie komórki. Analiza takich danych jest jednak niezwykle trudna: w każdej komórce można mierzyć poziomy aktywności dziesiątek tysięcy genów, z których wiele jest ledwie wykrywalnych. Istniejące modele AI upraszczają problem, skupiając się tylko na najsilniej aktywnych genach, co przyspiesza obliczenia, ale pomija wiele subtelnych sygnałów, które mogą być kluczowe w chorobie, rozwoju czy odpowiedzi na leki.
Nowy model, który słucha każdego genu
scLong podejmuje to wyzwanie, skalując się zamiast upraszczać. To model bazowy o miliardzie parametrów, trenowany na profilach aktywności genów pochodzących z około 48 milionów ludzkich komórek z ponad 50 tkanek. W przeciwieństwie do wcześniejszych podejść, które uwzględniały kilka tysięcy wysoko aktywnych genów, scLong rozważa jednocześnie około 28 000 genów, także tych rzadko lub słabo eksprymowanych. Łączy dwa rodzaje informacji dla każdego genu: jego poziom aktywności w danej komórce oraz to, co wiadomo o jego funkcji z Gene Ontology, obszernego, ekspercko kategoryzowanego katalogu ról i relacji genów. Specjalizowana sieć działająca na grafie połączeń między genami destyluje tę wiedzę a priori do zwartej reprezentacji, którą model może wykorzystać obok surowych wartości ekspresji.
Jak model równoważy moc i efektywność
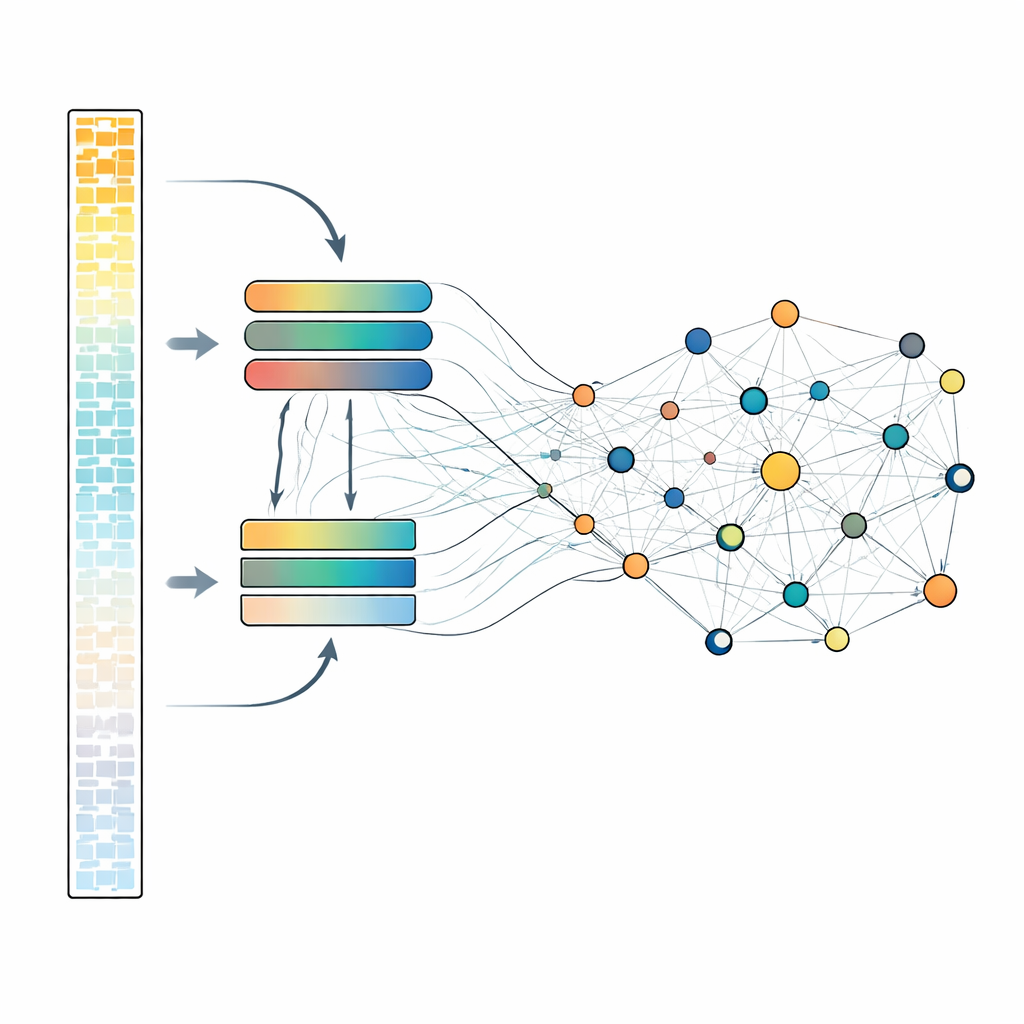
Szczegółowa analiza każdego genu jest kosztowna obliczeniowo, dlatego scLong korzysta z pomysłowej, dwuścieżkowej konstrukcji. W obrębie każdej komórki geny są sortowane według siły ekspresji. Najbardziej aktywne geny, które często niosą główny sygnał biologiczny, są przetwarzane przez większy, bardziej wydajny moduł uwagi. Cichsze geny, w tym pomiary niskie i zerowe, trafiają do mniejszego, lżejszego modułu. Następnie wszystkie geny są łączone i przekazywane przez kolejną warstwę uwagi, która pozwala każdemu genowi wpływać na pozostałe. Takie rozwiązanie pozwala modelowi zachować tańsze, lecz wciąż znaczące reprezentacje subtelnych sygnałów, jednocześnie rezerwując większą pojemność dla najsilniejszych z nich. Podczas wstępnego trenowania system wielokrotnie ukrywa podzbiór wartości aktywności genów i uczy się je rekonstruować na podstawie otaczającego kontekstu, zmuszając model do odkrywania wzorców łączących geny.
Wykorzystanie modelu w praktyce
Po treningu scLong można dostosować do szerokiego wachlarza pytań biologicznych. Autorzy pokazują, że model przewiduje, jak zmieni się aktywność genów, gdy konkretne geny zostaną wyłączone lub zmodyfikowane, w tym kombinacje dwóch genów działających wspólnie. Prognozuje także reakcję komórek na ekspozycję na różne związki chemiczne, co ma znaczenie dla odkrywania leków i testów bezpieczeństwa. W badaniach nad rakiem scLong pomaga przewidzieć, jak linie komórkowe nowotworu zareagują na pojedyncze leki oraz na pary leków, które mogą działać lepiej w kombinacji, często przewyższając zarówno wyspecjalizowane modele, jak i inne duże modele bazowe. Poza predykcją scLong potrafi wnioskować sieci regulacyjne między genami oraz pomagać korygować techniczne zniekształcenia powstające, gdy dane zbierane są w różnych laboratoriach lub na różnych urządzeniach.
Co to oznacza dla przyszłej medycyny i badań
Mówiąc prosto, scLong daje naukowcom mapę aktywności genów w pojedynczych komórkach o wysokiej rozdzielczości i świadomą kontekstu, która nie usuwa cichych ani rzadko używanych genów. Ucząc się na milionach komórek i integrując istniejącą wiedzę biologiczną, oferuje dokładniejsze przewidywania dotyczące reakcji komórek na zaburzenia genów, wprowadzenie nowych leków czy przebieg procesów chorobowych. Może to przyspieszyć poszukiwanie nowych terapii, wspierać bardziej spersonalizowane decyzje terapeutyczne i pogłębiać nasze rozumienie, jak złożone sieci genowe kontrolują zdrowie i chorobę. Choć model jest duży i wymaga znacznych zasobów obliczeniowych, wskazuje drogę ku przyszłości, w której potężne, ogólnego przeznaczenia systemy AI będą wszechstronnymi narzędziami do badania ukrytych mechanizmów komórek.
Cytowanie: Bai, D., Mo, S., Zhang, R. et al. scLong: a billion-parameter foundation model for capturing long-range gene context in single-cell transcriptomics. Nat Commun 17, 2380 (2026). https://doi.org/10.1038/s41467-026-69102-y
Słowa kluczowe: transkryptomika pojedynczych komórek, modele bazowe, regulacja genów, predykcja odpowiedzi na leki, ekspresja genów