Clear Sky Science · pl
Odkrywanie różnorodności PAM enzymu Cas9 poprzez metagenomiczne poszukiwania i uczenie maszynowe
Dlaczego to ma znaczenie dla przyszłej edycji genów
CRISPR stał się synonimem współczesnej edycji genów, lecz jedna dyskretna zasada nadal ogranicza jego możliwości: każde nacięcie w DNA musi znajdować się obok krótkiego „zezwolenia” w postaci sekwencji. Te krótkie wzorce, zwane PAM, decydują, gdzie popularna endonukleaza Cas9 może działać, a gdzie nie. Badanie to pokazuje, jak przeszukiwanie olbrzymich zasobów DNA mikroorganizmów, połączone z zaawansowanym uczeniem maszynowym, może ujawnić ogromną, ukrytą różnorodność tych „zezwoleń”. Nowa mapa może otworzyć znacznie więcej miejsc w ludzkim genomie dla precyzyjniejszych i bezpieczniejszych terapii.
Ukryte reguły kierujące nacięciami CRISPR
Cas9 i pokrewne enzymy są częścią naturalnego systemu odpornościowego występującego u bakterii i archeonów. Aby uniknąć cięcia własnego DNA, te mikroby sprawiają, że białka Cas poszukują PAM — bardzo krótkiego odcinka nukleotydów — obok miejsca docelowego. Dopiero gdy taki PAM jest obecny, Cas9 rozwija helisę DNA i pozwala przewodnikowi RNA sprawdzić dopasowanie, wywołując cięcie, jeśli wszystko się zgadza. Problem w medycynie polega na tym, że powszechnie stosowane w laboratoriach warianty, na przykład standardowy Cas9 z Streptococcus pyogenes, rozpoznają jedynie wąskie wzorce PAM. Jeśli mutacja wywołująca chorobę nie ma odpowiedniej sekwencji w pobliżu, dzisiejsze narzędzia po prostu nie sięgają po nią bez utraty precyzji.
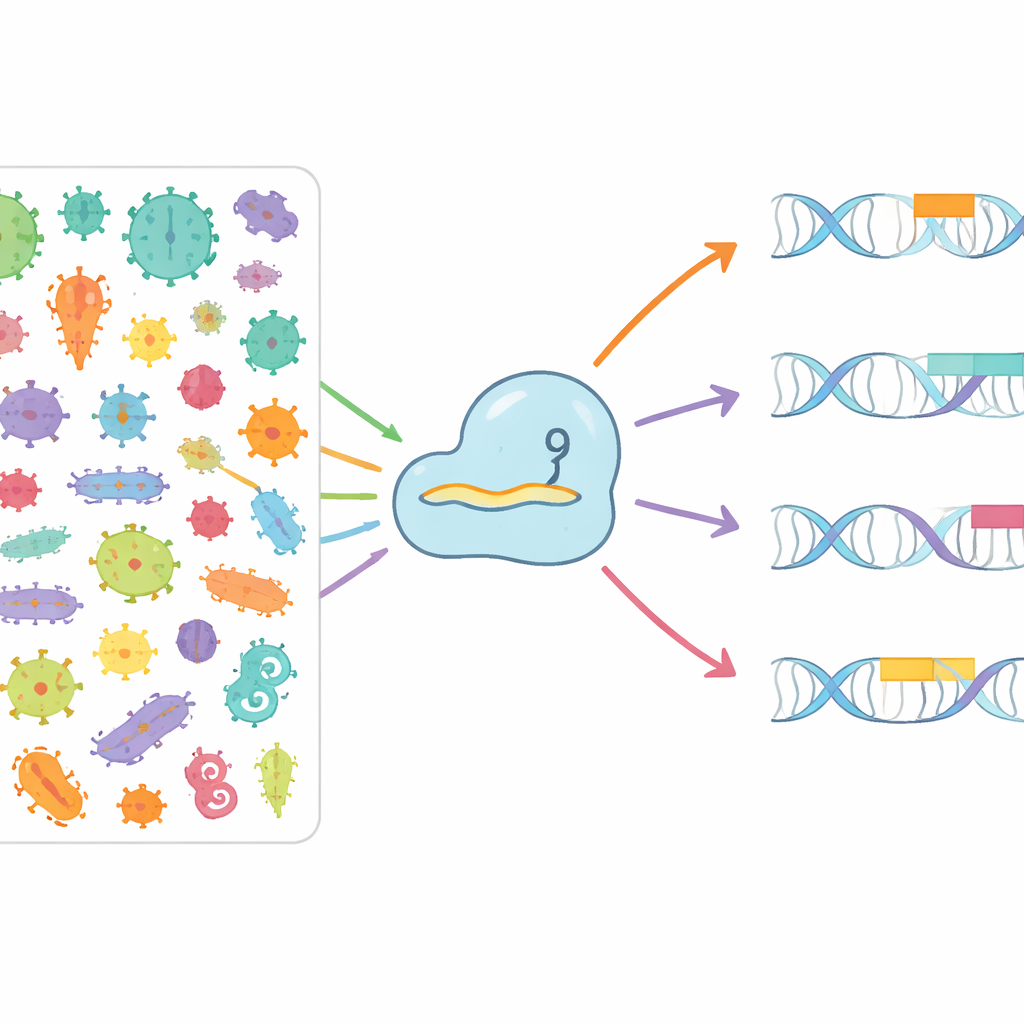
Poszukiwanie w świecie mikroorganizmów nowych opcji
Autorzy postawili sobie za cel systematyczne zmapowanie, jak różne białka Cas9 rozpoznają różne PAM w przyrodzie. Przeszukali ponad 3,8 miliona genomów bakteryjnych i archealnych oraz ponad 7,4 miliona sekwencji wirusowych i plazmidowych, które infekują lub przemieszczają się między mikroorganizmami. Poprzez identyfikację tablic CRISPR, łączenie ich z sąsiednimi genami Cas9 i dopasowywanie przechowywanych „pamięci” spacerów do atakujących wirusów i plazmidów, mogli zobaczyć, które krótkie wzorce DNA zwykle otaczają rzeczywiste cele. Na tej podstawie zbudowali CRISPR-PAMdb — publiczny katalog zawierający 8003 grup Cas9, z każdą sparowaną z konsensusowym profilem PAM, uporządkowany na drzewie ewolucyjnym, które uwypukla, jak blisko spokrewnione enzymy Cas9 mają tendencję do dzielenia podobnych preferencji PAM, przy jednoczesnym wykazywaniu zaskakującej ogólnej różnorodności.
Gdy danych brakuje, niech model się nauczy
Nawet przy tak rozległym przeglądzie, większość znalezionych białek Cas9 nie miała wystarczającej liczby dopasowanych celów wirusowych, by bezpośrednio odczytać PAM. Aby wypełnić luki, zespół opracował model uczenia maszynowego zwany CICERO. CICERO wykorzystuje potężny „model języka” białek, który nauczył się ogólnych wzorców sekwencji aminokwasowych i dostraja go do przewidywania, dla danego białka Cas9, jak prawdopodobne jest pojawienie się każdej z zasad DNA na każdej z dziesięciu pozycji w PAM. Model trenowano na profilach PAM z CRISPR-PAMdb, a następnie testowano zarówno przez walidację krzyżową, jak i na 79 enzymach Cas9, których PAMy zmierzono eksperymentalnie, osiągając silną zgodność między przewidywaniem a rzeczywistością.
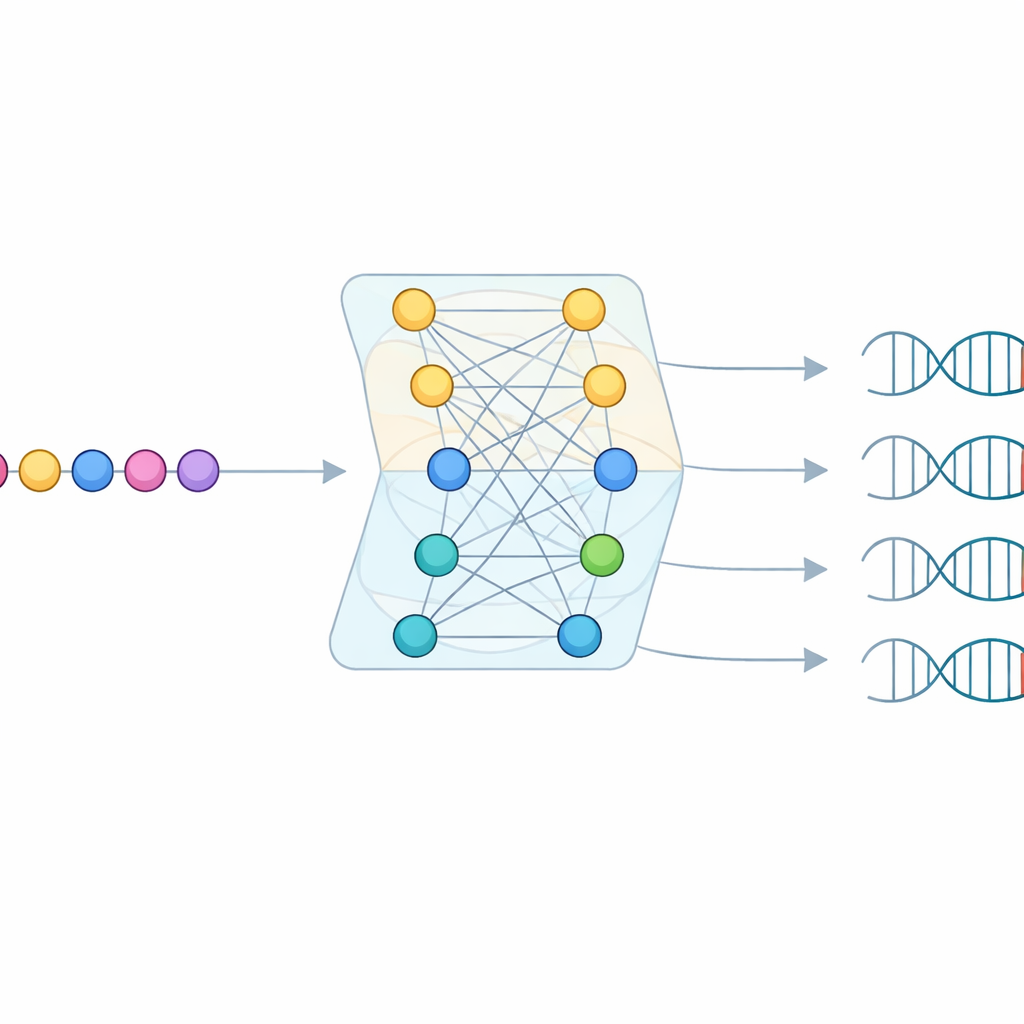
Wiedzieć, jak bardzo można ufać
Kluczową cechą CICERO jest to, że model nie tylko zgaduje PAM — szacuje też, jak wiarygodne jest każde przewidywanie. Po nauczeniu się przewidywania wzorców PAM, badacze wytrenowali drugą, lekką sieć, która przyjmuje tę samą sekwencję Cas9 i uczy się prognozować, jak dokładne będzie przewidywanie PAM. Wyższe wartości ufności silnie korelowały z wyższą rzeczywistą dokładnością. Dzięki temu filtrowi ufności zespół rozszerzył adnotacje PAM na ponad 50 000 dodatkowych białek Cas9, z ponad 17 000 przewidywań sklasyfikowanych jako wysokiego zaufania. To znacznie poszerza zestaw wariantów Cas9 o stosunkowo dobrze poznanych regułach celowania.
Co to oznacza dla leczenia chorób genetycznych
Aby pokazać praktyczne znaczenie tych nowych zasobów, autorzy przeanalizowali dziesiątki tysięcy pojedynczych mutacji literowych powiązanych z chorobami z bazy ClinVar, które w zasadzie można by skorygować za pomocą edytorów zasad — narzędzi zmieniających jedną literę DNA bez przecinania obu nici. Stwierdzili, że standardowy enzym Cas9 może uzyskać dostęp tylko do około połowy takich miejsc ze względu na swoje rygorystyczne wymagania dotyczące PAM. Gdy uwzględniono krewnych Cas9 z CRISPR-PAMdb oraz przewidywania CICERO o wysokim zaufaniu, które rozpoznają szerszy, ale nadal specyficzny zestaw pobliskich sekwencji, niemal wszystkie te mutacje stały się teoretycznie osiągalne bez rozluźniania zasad celowania w stopniu zagrażającym precyzji.
Większe narzędzie do precyzyjnej chirurgii DNA
Mówiąc prosto, ta praca tworzy dwa zasoby: ogromną, publiczną mapę łączącą tysiące naturalnych białek Cas9 z krótkimi wzorcami DNA, które preferują, oraz przewodnik AI, który potrafi przewidzieć te preferencje dla wielu kolejnych enzymów jedynie na podstawie ich sekwencji. Razem przekształcają świat mikroorganizmów w bogaty katalog części dla przyszłych edytorów genów. W miarę jak badacze będą dopracowywać i testować te warianty Cas9 w laboratorium, klinicyści mogą zyskać bezpieczniejsze, bardziej wszechstronne narzędzia, które będą mogły sięgać po mutacje wywołujące choroby, wcześniej niedostępne, przybliżając prawdziwie precyzyjną chirurgię genomu do praktycznej realizacji.
Cytowanie: Fang, T., Bogensperger, L., Feer, L. et al. Uncovering Cas9 PAM diversity through metagenomic mining and machine learning. Nat Commun 17, 2510 (2026). https://doi.org/10.1038/s41467-026-69098-5
Słowa kluczowe: CRISPR-Cas9, różnorodność PAM, metagenomika, uczenie maszynowe, edycja genomu