Clear Sky Science · pl
Predykcja podobieństwa chemicznego między tandemowymi widmami mas w różnych trybach jonizacji w metabolomice
Dlaczego łączenie chemicznych kropek ma znaczenie
Każdy łyk kawy, oddech powietrza czy dawka leku pozostawia w naszym organizmie śladowe ilości związków chemicznych. Nowoczesne przyrządy potrafią wykryć jednocześnie tysiące takich molekuł, lecz przekształcenie tych sygnałów w biologiczne wnioski wciąż bywa zaskakująco trudne. W tym badaniu przedstawiono MS2DeepScore 2.0 — narzędzie oparte na uczeniu maszynowym, które pomaga naukowcom dostrzec powiązania między tymi molekułami, nawet gdy sygnały są rejestrowane w bardzo różnych warunkach. Dzięki temu obiecuje szybszą i pełniejszą interpretację złożonych mieszanin chemicznych w medycynie, żywieniu i badaniach środowiskowych.
Dwa sposoby oglądania tej samej molekuły
Spektrometria mas to podstawowa technika, która waży i rozpada molekuły, aby ujawnić ich tożsamość. W rutynowych eksperymentach naukowcy często mierzą ten sam próbkę w dwóch trybach: jednym preferującym cząstki naładowane dodatnio, drugim — ujemnie. Każdy tryb daje charakterystyczny „kod kreskowy” fragmentów. Nawet gdy obie pomiary pochodzą od tej samej molekuły, powstałe wzorce mogą wyglądać tak różnie, że tradycyjne metody porównawcze zawodzą. W efekcie badacze zazwyczaj analizują tryby oddzielnie, budując dwie niepołączone mapy próbki i ryzykując przeoczenie istotnych zależności między związkami chemicznymi.
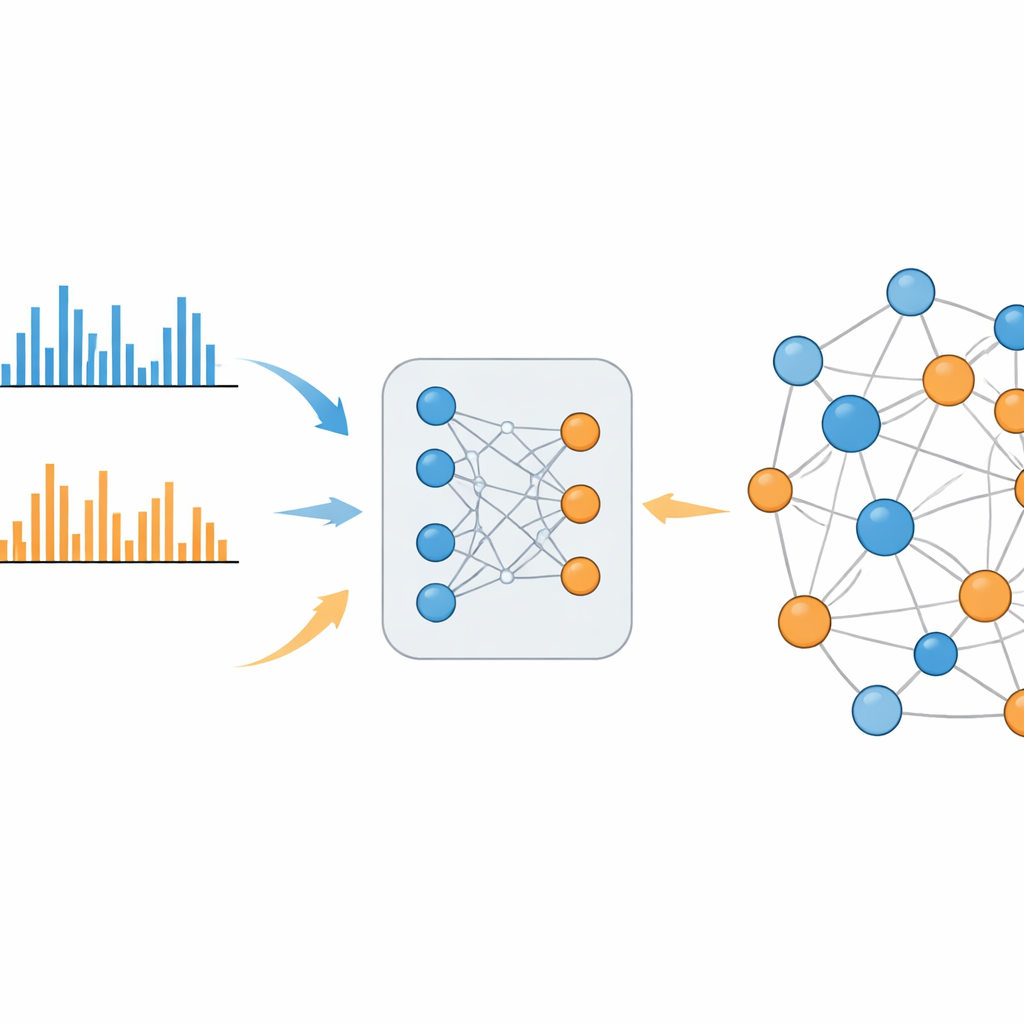
System uczący się, który wypełnia lukę
MS2DeepScore 2.0 rozwiązuje ten problem, ucząc się podobieństwa chemicznego bezpośrednio z dużych bibliotek znanych widm. Model opiera się na architekturze bliźniaczych sieci neuronowych, które przekształcają każdy wzorzec fragmentacji w 500-elementowy odcisk palca, zwany osadzeniem (embedding). Podczas treningu system widzi setki tysięcy przykładów z trybów dodatniego i ujemnego oraz informacje o rzeczywistym podobieństwie odpowiadających im molekuł. Dopasowuje swoje parametry tak, aby widma powiązanych molekuł miały podobne osadzenia, niezależnie od trybu pomiaru. Nowa wersja rozszerza poprzednika, wprowadzając dodatkowe informacje, takie jak masa pierwotnej molekuły i użyty tryb jonizacji, oraz stosując starannie zbalansowany schemat próbkowania, aby rzadkie, lecz informatywne relacje chemiczne nie zostały zagłuszone przez powszechne, mało informatywne przypadki.
Od rozproszonych sygnałów do zjednoczonych map
Po przeszkoleniu MS2DeepScore 2.0 potrafi oszacować chemiczne podobieństwo dowolnych dwóch widm, w tym par łączących tryby dodatni i ujemny. Autorzy wykazują, że te prognozy dobrze korelują z ustalonymi miarami podobieństwa strukturalnego — nie tylko w obrębie każdego trybu, lecz także między trybami. Korzystając z rzeczywistych danych z moczu ludzkiego, osocza krwi i dziko rosnącej jadalnej rośliny, zbudowali „sieci molekularne”, w których każde widmo jest węzłem, a silne przewidywane podobieństwo tworzy krawędzie. W przeciwieństwie do starszych podejść, te sieci naturalnie łączą dane z trybu dodatniego i ujemnego w jedną, spójną mapę. Klastery wyselekcjonowane przez ekspertów obejmują na przykład grupy związków powiązanych z kofeiną w moczu, które są powiązane między trybami i odpowiadają znanym szlakom metabolicznym.
Widzenie krajobrazu chemicznego na pierwszy rzut oka
Sieci molekularne są potężnym narzędziem, ale mogą się zaplątać, jeśli uwzględni się zbyt wiele słabych powiązań. Aby temu zapobiec, autorzy wykorzystują osadzenia MS2DeepScore bezpośrednio jako współrzędne w dwuwymiarowym układzie utworzonym techniką UMAP. Każda kropka na tej mapie reprezentuje jedno widmo, a bliskie kropki odpowiadają molekułom, które model uznaje za chemicznie podobne. Widma tego samego związku w trybach dodatnim i ujemnym — które wzrokowo wyglądają bardzo różnie — często pojawiają się obok siebie w tej przestrzeni osadzeń. Zespół wytrenował też dodatkowy model, który ocenia każde osadzenie pod kątem jego wiarygodności, oznaczając widma zaszumione, niekompletne lub niepodobne do niczego widzianego podczas treningu. Usunięcie tych punktów niskiej jakości poprawia ogólną dokładność i zwiększa zaufanie do wizualizacji.
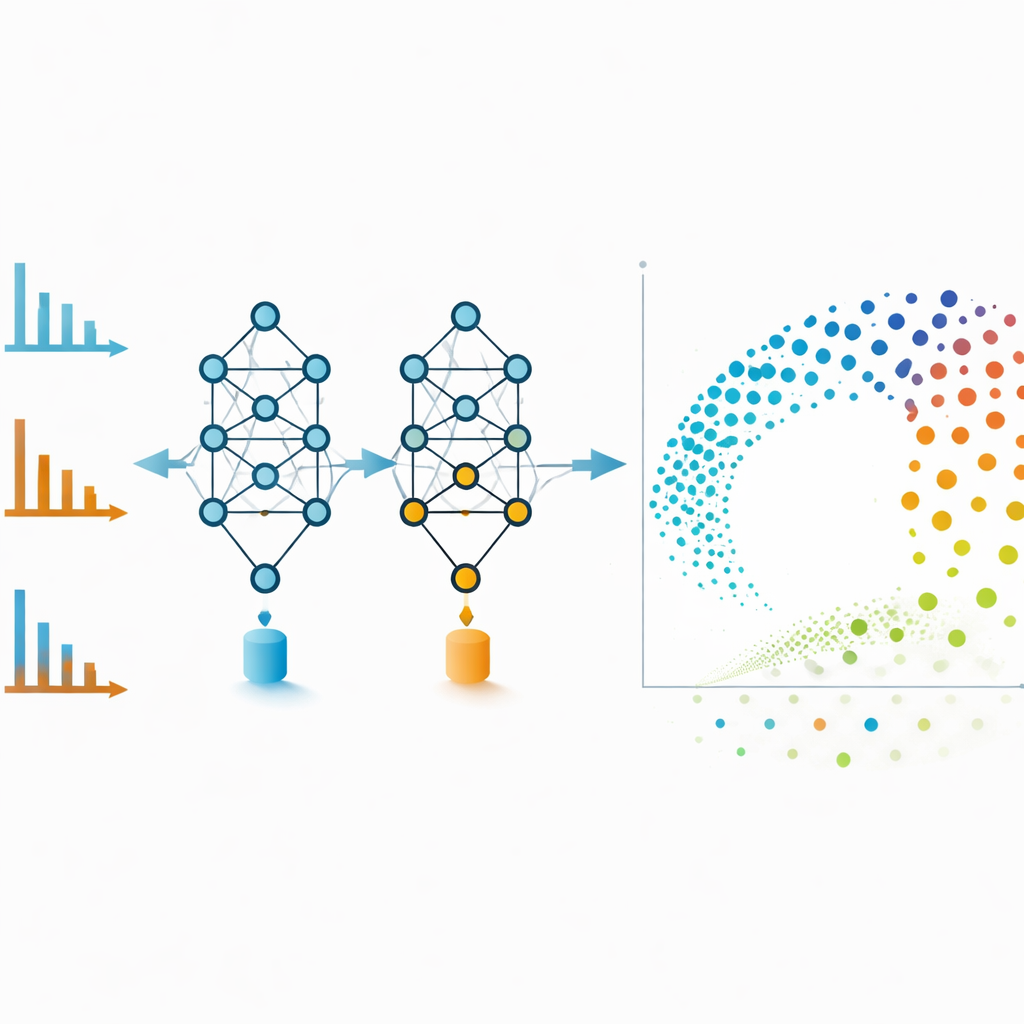
Udostępnianie zaawansowanych narzędzi laboratoriom na co dzień
Aby technologia była użyteczna także poza kręgiem programistów, autorzy zintegrowali MS2DeepScore 2.0 z popularnym, bezpłatnym oprogramowaniem do spektrometrii mas. Dzięki tej integracji badacze mogą wykrywać cechy, tworzyć sieci molekularne, które ignorują granice trybów jonizacji, oraz eksplorować powstałą przestrzeń chemiczną za pomocą interaktywnych pulpitów. Kod, wytrenowane modele i przykładowe zbiory danych są udostępnione otwarcie, a system można ponownie trenować lub dostroić pod wyspecjalizowane klasy związków chemicznych.
Co to oznacza dla przyszłych odkryć
Dla osób niebędących specjalistami kluczowy przekaz jest taki, że MS2DeepScore 2.0 pomaga przekształcić rozdrobnione i zależne od trybu pomiary w jedno, bardziej przejrzyste przedstawienie molekuł obecnych w próbce. Dzięki niezawodnemu łączeniu sygnałów, które wcześniej żyły w oddzielnych światach analitycznych, metoda pozwala naukowcom korzystać z dużo większych bibliotek referencyjnych, porównywać próbki bardziej kompleksowo i skupiać uwagę na istotnych skupiskach pokrewnych związków. To krzyżowe łączenie danych ma przyspieszyć identyfikację biomarkerów, składników odżywczych, produktów naturalnych i zanieczyszczeń, pogłębiając w efekcie nasze zrozumienie, jak chemia kształtuje zdrowie i środowisko.
Cytowanie: de Jonge, N.F., Chekmeneva, E., Schmid, R. et al. Cross ionization mode chemical similarity prediction between tandem mass spectra in metabolomics. Nat Commun 17, 2483 (2026). https://doi.org/10.1038/s41467-026-69083-y
Słowa kluczowe: metabolomika, spektrometria mas, uczenie maszynowe, sieciowanie molekularne, podobieństwo chemiczne