Clear Sky Science · pl
Ocena technologii atlasingu single‑cell ATAC‑seq za pomocą modelowania sekwencja→funkcja
Odczytywanie instrukcji komórki
Każda komórka w twoim ciele odczytuje tę samą DNA, a jednak komórki mózgowe, mięśniowe i odpornościowe zachowują się bardzo inaczej. Artykuł podejmuje podstawową zagadkę stojącą za tą różnorodnością: jak krótkie odcinki DNA zwane enhancerami działają jak przełączniki, włączając i wyłączając geny w określonych typach komórek. Autorzy pokazują, że nowe, tańsze technologie laboratoryjne mogą generować ogromne zbiory danych potrzebne do trenowania nowoczesnych modeli głębokiego uczenia, które czytają sekwencje DNA i przewidują, które enhancery są aktywne w jakich komórkach, przybliżając nas do rzeczywistego rozszyfrowania „gramatyki” regulacyjnej genomu.
Tworzenie map otwartego DNA w pojedynczych komórkach
Enhancery zwykle znajdują się w odcinkach DNA, które są bardziej otwarte i dostępne, co ułatwia wiązanie się z nimi białkom regulacyjnym. Technika zwana single‑cell ATAC‑seq mierzy, które części genomu są otwarte w tysiącach do setek tysięcy poszczególnych komórek jednocześnie, tworząc „atlas” dostępnego DNA w wielu typach komórek. Takie atlasy są idealnym paliwem dla modeli głębokiego uczenia, które jako wejście przyjmują surową sekwencję DNA i uczą się przewidywać, jak silnie każdy mały region działa jako enhancer w danym typie komórek. Do tej pory jednak większość takich atlasów opierała się na drogich, komercyjnych instrumentach, co rodzi pytanie, czy niskokosztowe, otwarte metody mogą dostarczyć danych treningowych o porównywalnej wartości dla tych modeli.
Otwartoźródłowa alternatywa dla platform komercyjnych
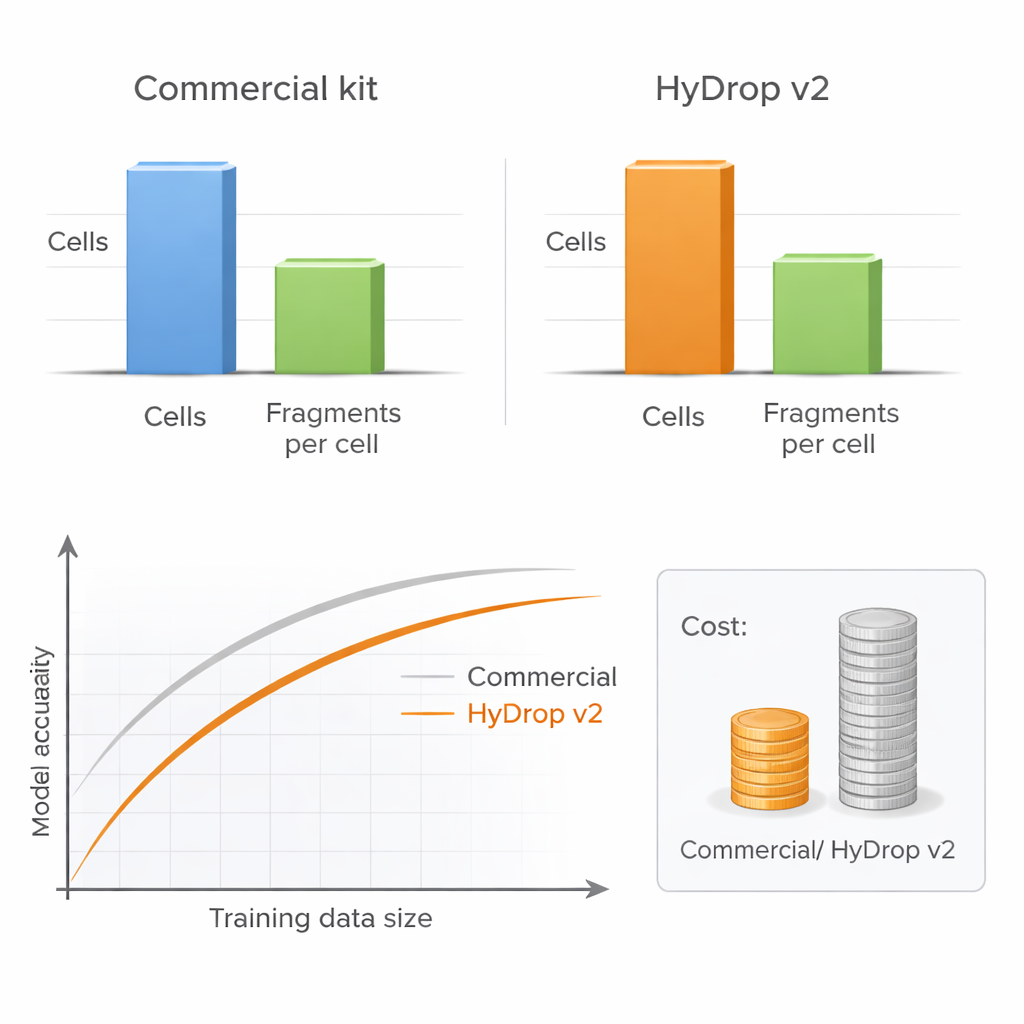
Autorzy przedstawiają HyDrop v2, ulepszoną metodę kroplową do single‑cell ATAC‑seq, która wykorzystuje niestandardowe hydrożelowe mikrokulki do znakowania kodami kreskowymi pojedynczych komórek. Porównują HyDrop v2 z powszechnie używanym zestawem komercyjnym, budując duże atlasy z dwóch bardzo różnych systemów: dorosłej kory ruchowej myszy i późnych stadiów embrionów muszki owocowej. HyDrop v2 generuje porównywalną jakość danych — odzyskując te same główne typy komórek i bardzo podobne zestawy dostępnych regionów DNA — przy koszcie około czternastokrotnie niższym na próbkę mózgu myszy. Co ważne, dane z eksperymentów HyDrop v2 integrują się płynnie z danymi komercyjnymi, co oznacza, że badacze mogą mieszać i dopasowywać platformy przy budowie bardzo dużych atlasów.
Trenowanie modeli głębokiego uczenia do odczytu logiki enhancerów
Aby sprawdzić, czy tańsze dane nadają się do zaawansowanego modelowania, zespół szkoli modele sekwencja→funkcja oparte na głębokim uczeniu na atlasach pochodzących z platform komercyjnych lub HyDrop v2. Modele te uczą się bezpośrednio z sekwencji DNA przewidywać, jak dostępny jest każdy region w każdym typie komórek, i potrafią wyróżniać krótkie wzorce sekwencyjne, które prawdopodobnie odpowiadają miejscom wiązania specyficznych białek regulacyjnych. W korze myszy modele trenowane na danych HyDrop v2 dorównują modelom opartym na danych komercyjnych pod względem ogólnej dokładności i zdolności do odzyskiwania znanych „przełączników” enhancerów, które wcześniej zostały zweryfikowane in vivo. W embrionie muszki obie platformy wspierają modele, które potrafią przeanalizować regiony o długości 2000 par zasad i wskazać kluczowe ~500‑par zasad segmenty, które faktycznie napędzają tkankowo‑specyficzną aktywność enhancerów, na przykład regiony kontrolujące ekspresję genów neuroblastów lub mięśni.
Więcej komórek potrafi przewyższyć większą głębokość
Kluczowe praktyczne pytanie dla każdego laboratorium brzmi: czy sekwencjonować każdą komórkę bardzo głęboko, czy profilować więcej komórek przy mniejszej głębokości. Poprzez systematyczne zmienianie liczby komórek i liczby fragmentów DNA na komórkę, autorzy pokazują, że wydajność modeli prawie nie ucierpi, gdy głębokość sekwencjonowania zostanie zredukowana do umiarkowanego poziomu, pod warunkiem że uwzględniona zostanie wystarczająca liczba komórek. W przeciwieństwie do tego, zmniejszenie liczby komórek wyraźnie pogarsza dokładność modelu, szczególnie gdy mierzy się wydajność w wielu typach komórek naraz. Ponieważ HyDrop v2 jest znacznie tańszy na komórkę, badacze mogą łatwo dodać dziesiątki tysięcy dodatkowych komórek, odzyskując lub nawet przewyższając wydajność modeli opartych na danych komercyjnych za ułamek kosztów.
Widzenie odcisków białek na DNA
Badanie sprawdza również, czy różne platformy laboratoryjne wprowadzają subtelne uprzedzenia w sposobie, w jaki enzym ATAC‑seq tnie DNA, co mogłoby wprowadzać w błąd modele próbujące wywnioskować, gdzie białka siedzą na genomie. Korzystając z oddzielnego narzędzia opartego na sieci neuronowej korygującego preferencje enzymu, autorzy pokazują, że HyDrop v2 i zestawy komercyjne generują niemal identyczne wzorce aktywności enzymu zarówno w komórkach myszy, jak i muszki. Po korekcji oba zbiory danych ujawniają drobnoskalowe „odciski”, gdzie białka regulacyjne i nukleosomy wydają się chronić DNA przed cięciem, a te odciski pokrywają się ze wzorcami sekwencyjnymi wyróżnionymi przez modele sekwencja→funkcja. Ta zgodność sugeruje, że platformy otwartoźródłowe i komercyjne są równie odpowiednie do szczegółowych badań nad tym, jak białka oddziałują z DNA.
Dlaczego to ma znaczenie dla rozszyfrowania genomu
Dla osób spoza specjalności główne przesłanie jest takie, że teraz możemy budować bardzo duże, przystępne cenowo mapy wykorzystania DNA w pojedynczych komórkach i szkolić potężne modele głębokiego uczenia na tych mapach bez polegania wyłącznie na kosztownym, zastrzeżonym sprzęcie. HyDrop v2 dostarcza danych wspierających przewidywanie enhancerów, interpretację wzorców sekwencyjnych i odciski wiązania białek na poziomie porównywalnym z wiodącymi metodami komercyjnymi, pod warunkiem przeprofilowania wystarczającej liczby komórek. To otwiera drogę do tworzenia atlasów elementów regulacyjnych obejmujących całe organizmy w zdrowiu i chorobie, przyspieszając wysiłki na rzecz odczytania instrukcji regulacyjnych genomu oraz projektowania nowych, precyzyjnie ukierunkowanych przełączników genetycznych do badań i przyszłych terapii.
Cytowanie: Dickmänken, H., Wojno, M., Mahieu, L. et al. Evaluating single-cell ATAC-seq atlasing technologies using sequence-to-function modeling. Nat Commun 17, 1951 (2026). https://doi.org/10.1038/s41467-026-68742-4
Słowa kluczowe: single‑cell ATAC‑seq, enhancery, modele głębokiego uczenia, regulacja genów, otwarta genomika