Clear Sky Science · pl
Rzetelne przewidywanie numerów Komisji Enzymów za pomocą hierarchicznego interpretowalnego transformera
Dlaczego przewidywanie „zadań” enzymów ma znaczenie
Każda żywa komórka działa dzięki niezliczonym, drobnym maszynom chemicznym zwanym enzymami. Każdy enzym ma określone „zadanie”, które zapisane jest jako numer Komisji Enzymów (EC) — czteroczęściowy kod podobny do adresu pocztowego. Poprawne przypisywanie numerów EC jest kluczowe dla zrozumienia metabolizmu, projektowania nowych leków, inżynierii mikroorganizmów produkujących paliwa czy alternatywy dla tworzyw sztucznych oraz śledzenia, jak ekosystemy przetwarzają związki chemiczne. Doświadczenia określające funkcje enzymów są jednak powolne i kosztowne. W tym badaniu przedstawiono HIT-EC, nowy model sztucznej inteligencji, który potrafi wiarygodnie przewidywać numery EC na podstawie sekwencji białkowych, jednocześnie wyjaśniając, dlaczego podjął daną decyzję.
System przypominający kod pocztowy dla zadań enzymów
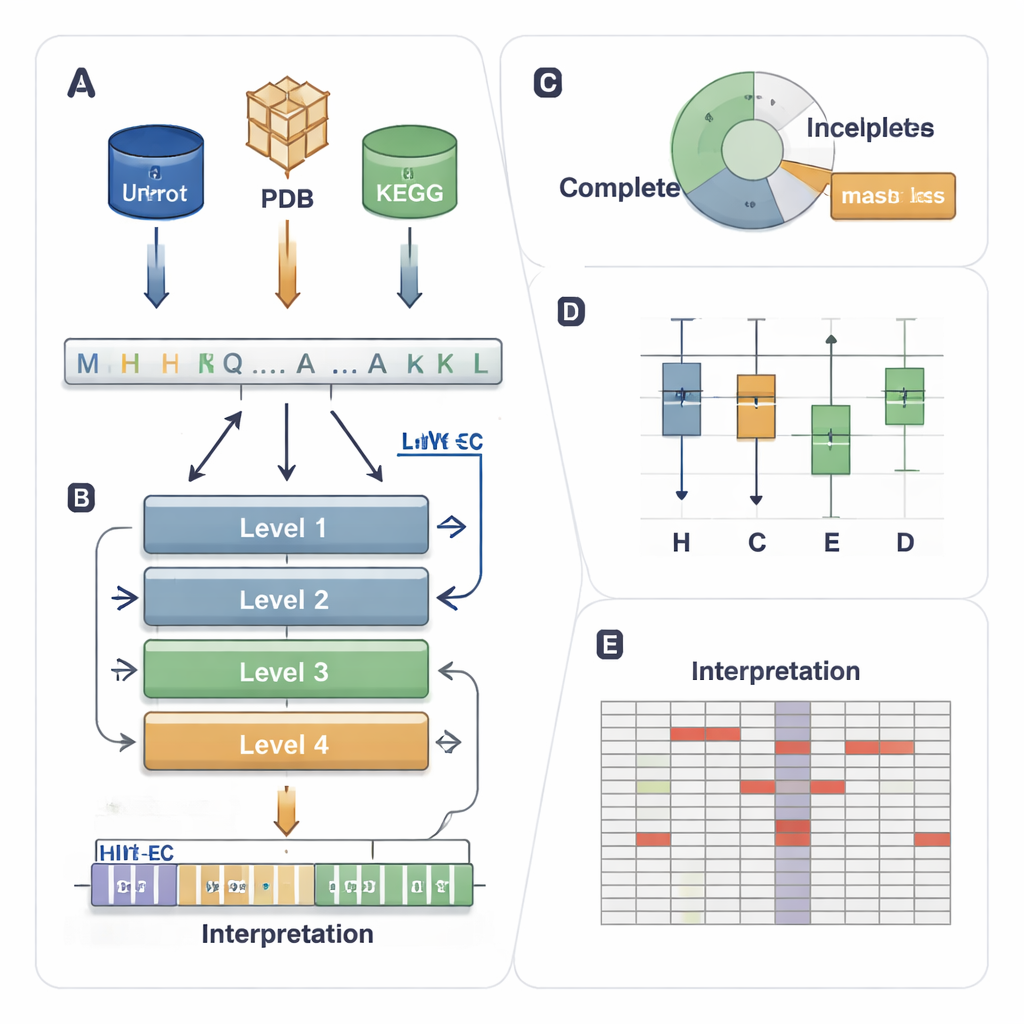
System EC przypisuje każdemu enzymowi czteropoziomowy kod, na przykład 1.1.1.37. Pierwsza cyfra wskazuje szeroką klasę (np. enzymy przenoszące elektrony lub grupy), a kolejne cyfry opisują coraz dokładniejsze szczegóły reakcji. Ta hierarchia jest potężna, ale stawia trudne wymagania przy przewidywaniu: model musi poprawnie odgadnąć wszystkie cztery poziomy spośród tysiąca możliwych kodów, nawet gdy niektóre enzymy są rzadkie lub częściowo oznakowane w bazach danych (np. 3.5.-.-, gdzie brak jest szczegółowych poziomów). Istniejące metody komputerowe korzystają z struktury 3D, podobieństwa sekwencji lub uczenia głębokiego, ale mają trudności z rzadkimi enzymami, pomijają dane częściowo oznakowane i zwykle działają jako „czarne skrzynki”, które niewiele mówią o przyczynach swoich predykcji.
AI o czterech kondygnacjach, podążająca po drabinie EC
HIT-EC (Hierarchical Interpretable Transformer for EC prediction) zaprojektowano tak, by odzwierciedlał czterostopniową hierarchię EC. Przyjmuje surową sekwencję białkową i przepuszcza ją przez cztery warstwy transformera, z których każda koncentruje się na jednym poziomie EC. Lokalne przepływy łączą każdy poziom z poprzednim, zapewniając, że decyzja szczegółowa (czwarta cyfra) jest zgodna z decyzjami o szerszym zasięgu (pierwsza i druga cyfra). Równolegle globalny przepływ utrzymuje kontekst całej sekwencji na każdym etapie. Model można także trenować na sekwencjach z brakującymi etykietami, używając „zamaskowanej straty”, która po prostu ignoruje brakujące poziomy EC zamiast odrzucać sekwencję. Dzięki temu HIT-EC uczy się na dużej części białek w bazach kuration, które są jedynie częściowo oznakowane.
Przewyższając konkurencję pod względem dokładności i prędkości
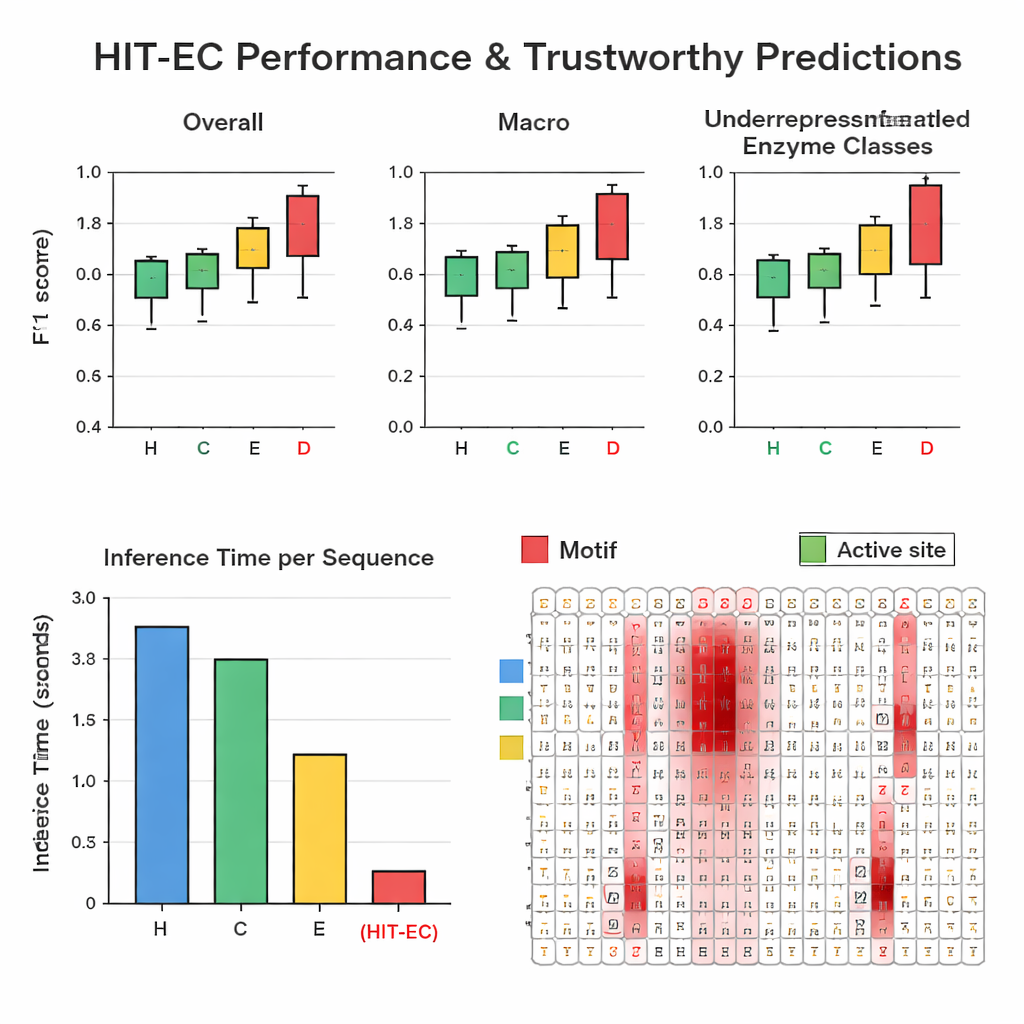
Autorzy zebrali dużą, starannie przefiltrowaną bazę danych liczącą około 200 000 enzymów z 1 938 różnymi numerami EC, pochodzącą ze Swiss-Prot i Protein Data Bank. W powtarzanych testach z podziałem na zestaw treningowy i testowy HIT-EC pokonał trzy wiodące metody (CLEAN, ECPICK i DeepECtransformer) zarówno w ogólnych, jak i per-klasowych miarach F1, które oceniają równowagę między trafieniami a fałszywymi alarmami. Szczególnie dobrze radził sobie z rzadko reprezentowanymi kodami EC mającymi 25 lub mniej znanych przykładów, gdzie wcześniejsze metody często zawodzą. HIT-EC dobrze uogólniał również na nowe enzymy dodane do Swiss-Prot po treningu oraz na pełne genomy różnych bakterii, w tym dobrze zbadanych szczepów Escherichia coli, Bacillus subtilis i Mycobacterium tuberculosis. Pomimo zaawansowania model był bardzo wydajny: na standardowym GPU przetwarzał białko w około 38 milisekund — dziesiątki razy szybciej niż niektóre konkurencyjne podejścia zależne od wolniejszych wyszukiwań podobieństwa lub zespołów wielu modeli.
Pokazywanie, „na co” patrzy model
Aby uczynić przewidywania wiarygodnymi, HIT-EC zaprojektowano tak, by ujawniał, które aminokwasy w sekwencji wpłynęły na decyzję na danym poziomie EC. Autorzy opracowali ścieżkę interpretacyjną łączącą wagi uwagi z informacją gradientową, aby ocenić ważność każdej pozycji. Zweryfikowali te oceny na dobrze scharakteryzowanych rodzinach enzymów. Na przykład w rodzinie cytochromów P450 (CYP106A2) HIT-EC wyróżnił znane motywy funkcjonalne, takie jak regiony wiążące tlen i hem, i zidentyfikował subtelny motyw EXXR, który umknął jednemu z modeli referencyjnych. Dla klasycznych przedstawicieli każdej najwyższej klasy EC — takich jak dehydrogenaza alkoholowa, heksokinaza czy anhydraza węglanowa — oceny relewantności modelu podświetlały podręcznikowe motywy sygnaturowe i miejsca wiążące substraty. Te interpretacje dostarczają biochemicznego „dowodu”, że model opiera swoje decyzje na znaczących cechach, a nie na przypadkowych korelacjach.
Wskazówki dla badań nad rzadkimi i pojawiającymi się enzymami
Zespół dodatkowo przetestował HIT-EC na dwóch słabo zbadanych enzymach ważnych dla oczyszczania zanieczyszczeń: cytochromie P450 zaangażowanym w rozkład aromatycznych zanieczyszczeń oraz hydrolazie rozkładającej PET ze Streptomyces, która pomaga trawić cząsteczki związane z plastikiem. Oba enzymy były scharakteryzowane eksperymentalnie, lecz nie miały oficjalnych przypisań EC. HIT-EC poprawnie przewidział oczekiwane numery EC i wyróżnił wzorce motywów oraz reszty katalityczne zgodne z tym, co wiadomo ze studiów strukturalnych i biochemicznych. Podsumowując, praca pokazuje, że HIT-EC może nie tylko przypisywać numery EC dokładniej i szybciej niż obecne narzędzia — zwłaszcza dla rzadkich funkcji — ale także wyjaśniać, dlaczego dany enzym uważa się za wykonujący określone zadanie chemiczne. To połączenie wydajności i interpretowalności czyni go obiecującym narzędziem do masowej, wiarygodnej adnotacji enzymów w genomice, biotechnologii i badaniach środowiskowych.
Cytowanie: Dumontet, L., Han, SR., Lee, J.H. et al. Trustworthy prediction of enzyme commission numbers using a hierarchical interpretable transformer. Nat Commun 17, 1146 (2026). https://doi.org/10.1038/s41467-026-68727-3
Słowa kluczowe: predykcja funkcji enzymów, uczenie głębokie w biologii, modele transformerowe, adnotacja białek, enzymy do bioremediacji