Clear Sky Science · pl
Alternatywna aktywacja EGFR przez mutację R252C pochodzącą od pacjenta wspiera postęp choroby nowotworowej
Kiedy anteny komórkowe wariują
Dlaczego niektóre nowotwory nadal rosną pomimo serii chemioterapii i nowoczesnej immunoterapii? Badanie śledzi pacjenta z guzami w płucach i mózgu i odtwarza przebieg choroby aż do drobnej zmiany w kluczowej „antennie” na powierzchni komórki zwanej EGFR. Odkrywając, jak ta pojedyncza mutacja przekańcza sygnały wzrostu na nowo, badacze wyjaśniają agresywny przebieg choroby u pacjenta i pokazują, że istniejący lek — afatinib — może go powstrzymać.
Rzadka mutacja o dużych konsekwencjach
EGFR to receptor zakotwiczony w błonie komórkowej, który pomaga komórkom reagować na sygnały wzrostowe. Wiele raków płuca i mózgu niesie mutacje w EGFR, ale większość znanych zmian występuje wewnątrz komórki, w części działającej jak przełącznik enzymatyczny. W tym badaniu zespół odkrył nietypową zmianę na zewnątrz EGFR, w fragmencie, który normalnie łapie czynniki wzrostu. U pacjenta z rakiem płuca i glejakiem stwierdzono zamianę jednego aminokwasu na pozycji 252 — zamiast argininy pojawiła się cysteina — oznaczoną jako EGFR R252C. Przeszukanie baz danych nowotworowych wykazało tę mutację u niewielkiej części pacjentów z glejakiem i prawie nigdy w guzach płuca, co sugeruje, że jest rzadka, ale rzeczywista. Za pomocą narzędzi do edycji genów autorzy odtworzyli tę dokładną mutację w kilku liniach komórek nowotworowych mózgu i płuca, aby zbadać jej skutki.
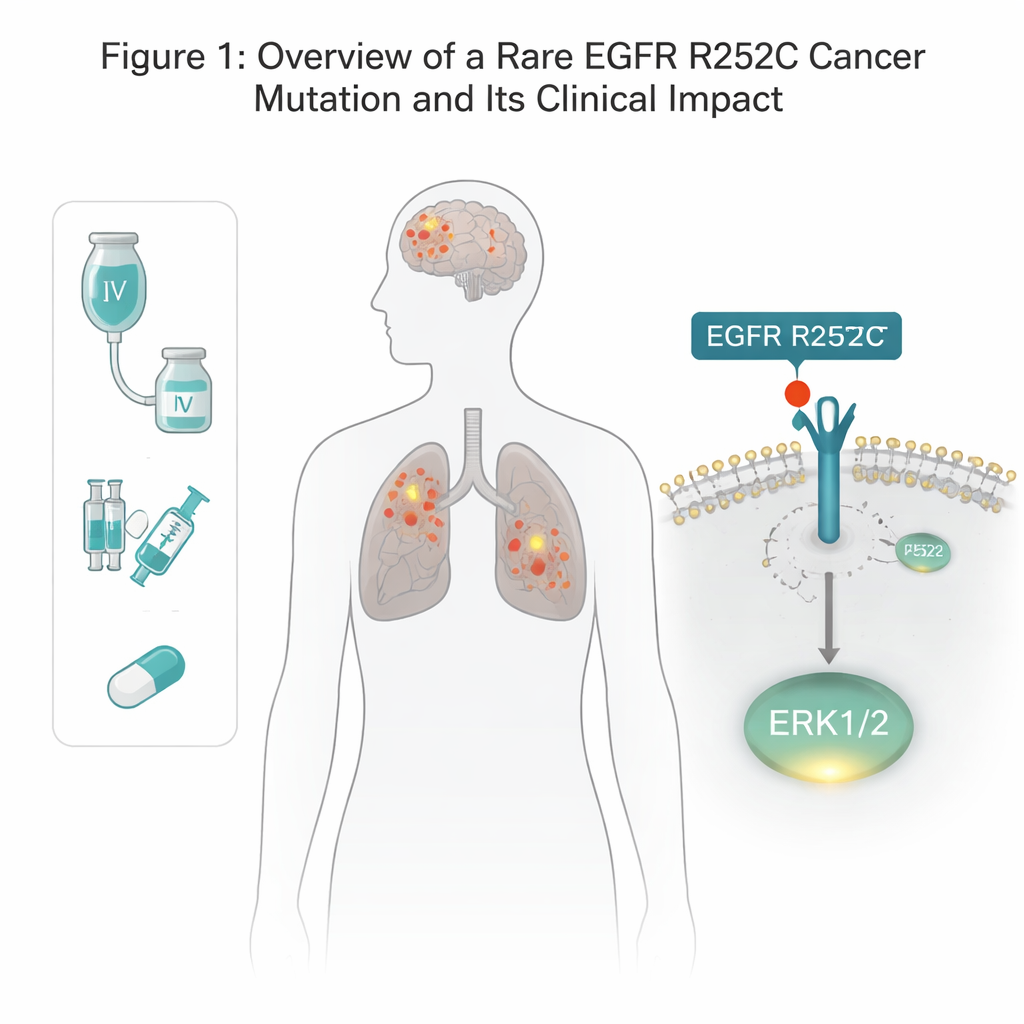
Nowe skróty do sygnałów wzrostu
Zwykle EGFR musi utworzyć parę z drugim egzemplarzem i następnie oznakować swoją wewnętrzną końcówkę grupami fosforanowymi, zanim uruchomi szlaki wzrostowe. Co zaskakujące, wersja R252C nie wykazywała tego zwykłego samoznakowania. Zamiast tego komórki z EGFR R252C silniej niż normalnie aktywowały konkretny regulator wzrostu — ERK1/2 — przy utrzymaniu innych klasycznych szlaków EGFR, takich jak AKT i STAT3, w dużej mierze bez zmian. Zablokowanie ERK1/2 za pomocą specyficznego inhibitora likwidowało dodatkową przewagę wzrostową komórek R252C, dowodząc, że ERK1/2 jest głównym silnikiem napędzającym nowotworową aktywność tej mutacji.
Zablokowanie receptora w stanie ciągłej aktywności
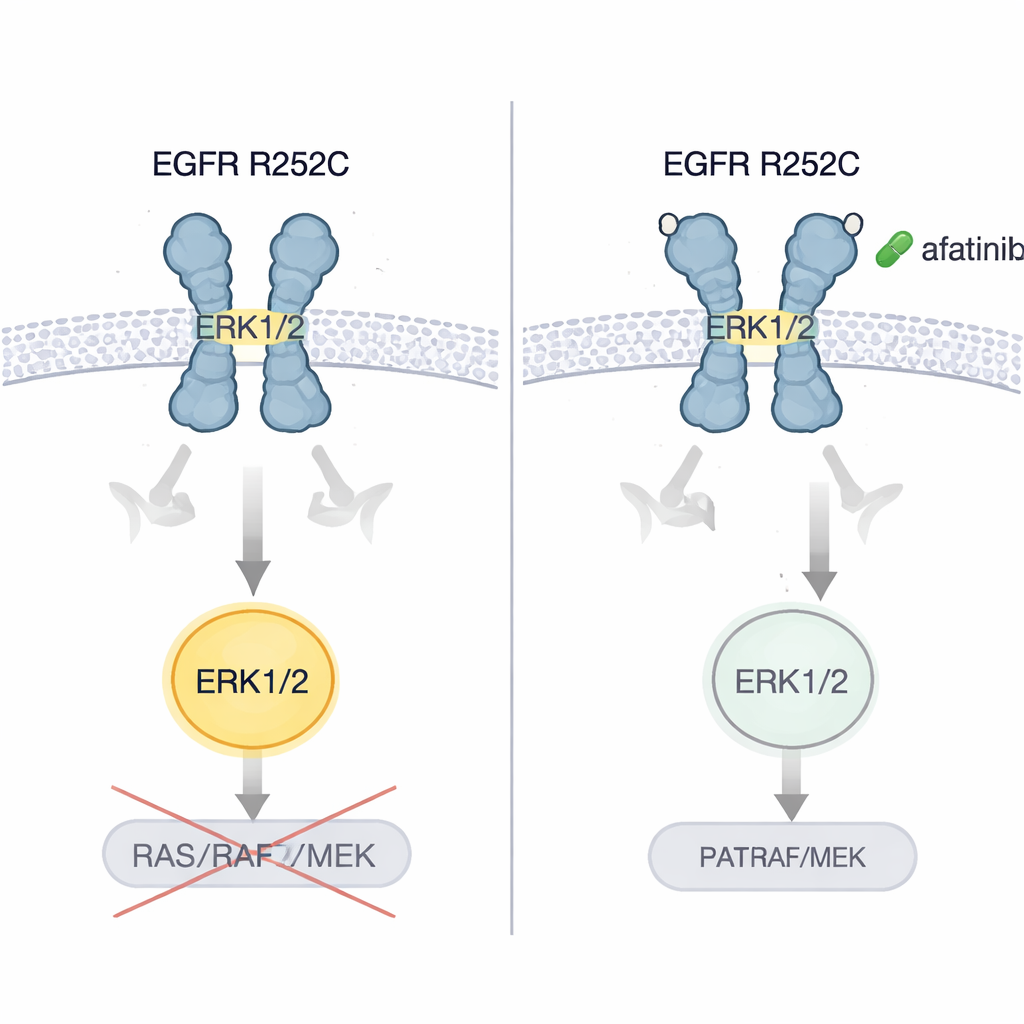
Aby zrozumieć, jak zmiana zewnętrzna może powodować tak wybiórcze nadaktywowanie, badacze połączyli testy biochemiczne z symulacjami komputerowymi. Zamiana na R252C wprowadza nową cysteinę w zewnętrznej części EGFR. Dwa takie mutanty mogą utworzyć mostek dwusiarczkowy — rodzaj molekularnego zszywacza — między resztami C252, trwale je łącząc w stabilną parę. Modelowanie strukturalne wykazało, że ten mostek wymusza na zewnętrznej części receptora „V-kształtne”, przesunięte ustawienie, które bardzo przypomina aktywną, związana z ligandem formę, nawet gdy czynnik wzrostu nie jest obecny. To ustawienie przenosi się przez segmenty przechodzące przez błonę i tuż wewnątrz niej, skręcając wewnętrzne domeny enzymatyczne w nietypową konfigurację: miejsca aktywne skierowane są w stronę wnętrza komórki, ale są zbyt oddalone, by efektywnie fosforylować się nawzajem. Zamiast tego ta konformacja tworzy silną powierzchnię dokującą dla ERK1/2, pozwalając EGFR R252C bezpośrednio fosforylować ERK1/2 i ominięć zwykłe połączenie RAS–RAF–MEK.
Od modeli mysich do jednego pacjenta
Autorzy wykazali, że komórki nowotworowe mózgu i płuca niosące EGFR R252C rosły szybciej in vitro i tworzyły większe, bardziej agresywne guzy po wszczepieniu do myszy, w porównaniu z komórkami z normalnym EGFR. Przetestowali kilka generacji leków blokujących EGFR. Tylko afatinib, inhibitor drugiej generacji, konsekwentnie wyłączał aktywację ERK1/2 i zdecydowanie ograniczał wzrost komórek nowotworowych. W modelach mysich guzów mózgu i płuca napędzanych przez R252C afatinib spowalniał ekspansję guza i wydłużał przeżycie. Co istotne, gdy pierwotny pacjent — u którego choroba pogarszała się pomimo chemioterapii, leku przeciwko naczyniom krwionośnym i immunoterapii — został przełączony na afatinib, badania obrazowe płuc i mózgu pokazały wyraźne zmniejszenie masy guza, a pacjent przez kilka lat nie miał progresji.
Co to oznacza dla pacjentów
Praca ujawnia wcześniej nierozpoznany sposób działania mutacji onkogennych EGFR: zszywanie dwóch receptorów na zewnątrz komórki, skręcanie ich w aktywną pozycję, która bezpośrednio włącza ERK1/2 zamiast podążać podręcznikowym kaskadowym szlakiem sygnałowym. Dla nie-specjalistów kluczowy wniosek jest taki, że nie wszystkie mutacje w tym samym genie zachowują się jednakowo, i że niektóre rzadkie zmiany mogą najlepiej reagować na konkretne istniejące leki. EGFR R252C wydaje się tworzyć nowotwory szczególnie wrażliwe na afatinib. Choć wnioski te opierają się obecnie na jednym szczegółowym przypadku pacjenta oraz szerokich badaniach laboratoryjnych, wskazują na potrzebę bardziej spersonalizowanego badania mutacji w zewnętrznych domenach EGFR i sugerują, że starannie dobrane terapie celowane mogą dać nową nadzieję wybranym pacjentom z trudnymi do leczenia guzami mózgu i płuca.
Cytowanie: Zhang, Y., Fei, Q., Li, Y. et al. An alternative EGFR activation by patient-derived R252C mutation promotes cancer progression. Nat Commun 17, 1902 (2026). https://doi.org/10.1038/s41467-026-68699-4
Słowa kluczowe: mutacja EGFR, glejak, rak płuca, szlak ERK, afatinib