Clear Sky Science · pl
Poprawa predykcji ocen poligenicznych dla grup niedostatecznie reprezentowanych poprzez uczenie transferowe
Dlaczego twój genetyczny wynik ryzyka może nie działać dla ciebie
Genetyczne „wskaźniki ryzyka” są coraz częściej używane do oszacowania szansy danej osoby na rozwój powszechnych schorzeń, takich jak cukrzyca, choroba serca czy nadciśnienie. Jednak większość tych wskaźników została zbudowana na danych DNA pochodzących od osób o europejskim pochodzeniu. W efekcie często źle sprawdzają się u osób z innych środowisk, co rodzi obawy o sprawiedliwość i użyteczność w medycynie. W tym badaniu postawiono proste pytanie: czy możemy ponownie wykorzystać wiedzę zgromadzoną w dużych europejskich zbiorach danych, aby zbudować lepsze, bardziej sprawiedliwe genetyczne wskaźniki dla grup niedoreprezentowanych — bez udostępniania surowych danych genetycznych?
Od milionów markerów DNA do pojedynczego wyniku ryzyka
Poligeniczny wynik to coś w rodzaju świadectwa, które sumuje drobne efekty wielu markerów genetycznych rozmieszczonych w całym genomie. Każdemu markerowi przypisywana jest waga odzwierciedlająca, jak silnie wiąże się z daną cechą, na podstawie dużych badań genetycznych. Gdy te badania dotyczą głównie Europejczyków, otrzymany wynik zazwyczaj najlepiej działa u osób pochodzenia europejskiego. Różnice w tle genetycznym — w tym częstotliwość niektórych wariantów i to, jak są współdziedziczone — sprawiają, że te same wagi często zawodzą w populacjach afroamerykańskich, latynoskich i innych. Zbieranie równie dużych zestawów danych dla każdej grupy jest kosztowne i wolne, więc autorzy sięgnęli po strategię uczenia maszynowego zwaną uczeniem transferowym: zamiast zaczynać od zera dla każdej populacji, dopracowują istniejący model wytrenowany gdzie indziej.
Jak pożyczać wiedzę bez udostępniania surowych danych
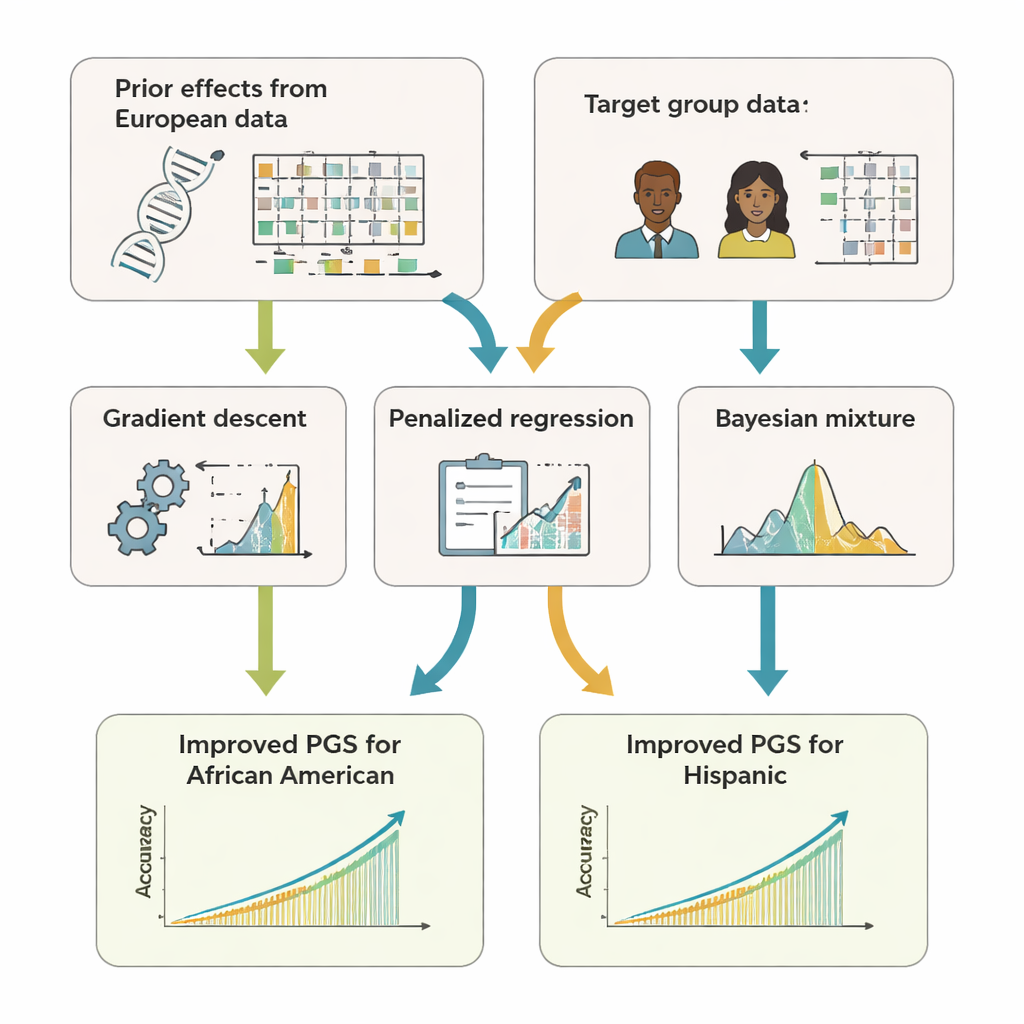
Zespół opracował GPTL, otwartoźródłowy pakiet w R, który implementuje trzy podejścia uczenia transferowego dla ocen genetycznych. Wszystkie trzy zaczynają od istniejących estymat efektów DNA uzyskanych w dużym zbiorze osób o europejskim pochodzeniu, a następnie delikatnie korygują te estymaty przy użyciu danych z grupy docelowej, takiej jak Afroamerykanie czy Latynosi. Jedna metoda modyfikuje europejskie wagi krok po kroku za pomocą spadku gradientu i zatrzymuje się wcześnie, zanim całkowicie je nadpisze. Druga metoda, zwana regresją z karą, aktywnie przyciąga nowe estymaty w stronę wartości oryginalnych, o ile dane grupy docelowej nie dostarczają silnych dowodów przeciwnych. Trzecia, mieszana metoda bayesowska, pozwala każdemu markerowi DNA wybrać spośród kilku źródeł informacji — na przykład wielu grup pochodzeniowych lub nawet opcji „brak efektu” — i miesza je zgodnie z tym, jak dobrze wyjaśniają dane grupy docelowej.
Testowanie metod w praktyce
Aby sprawdzić skuteczność tych podejść, autorzy użyli zarówno symulacji komputerowych, jak i rzeczywistych danych od setek tysięcy ochotników z UK Biobank oraz amerykańskiego programu badawczego All of Us. Skoncentrowali się na uczestnikach afroamerykańskich i latynoskich jako grupach docelowych, wykorzystując dane osób o europejskim pochodzeniu jako główne źródło informacji priorytetowej. W przypadku 11 cech — w tym wzrostu, wskaźnika masy ciała, lipidów krwi, ciśnienia krwi i markerów nerek — wskaźniki oparte na uczeniu transferowym konsekwentnie przewyższały wyniki zbudowane wyłącznie w grupie docelowej lub po prostu ponownie użyte z Europy. Często ich trafność równała się lub nieco przewyższała bardziej złożone metody „wielopochodzeniowe”, które wymagają łączenia surowych danych z kilku populacji. Co kluczowe, metody GPTL potrzebują jedynie statystyk sumarycznych — zagregowanych liczb opisujących efekty genetyczne — dzięki czemu instytucje mogą współpracować, nie ujawniając danych genetycznych na poziomie indywidualnym.
Kiedy więcej DNA nie zawsze znaczy lepiej
Naukowcy przeanalizowali także, jak najlepiej wybierać markery genetyczne do uwzględnienia. Wbrew powszechnemu przekonaniu, że użycie każdego dostępnego markera zawsze pomaga, stwierdzili, że dla grup afroamerykańskich, a szczególnie latynoskich, włączenie milionów bardzo słabych sygnałów może faktycznie pogorszyć wydajność, zwłaszcza przy użyciu silnie uproszczonych reprezentacji korelacji genetycznych. Skupienie się na lepiej udokumentowanych markerach i stosowanie bogatszych informacji o współdziedziczeniu wariantów często przynosiło bardziej trafne wyniki. Badanie wykazało również, że dodanie informacji priorytetowych z wielu grup pochodzeniowych i staranne modelowanie różnic między populacjami dodatkowo poprawia predykcję.
Co to oznacza dla bardziej sprawiedliwej predykcji genetycznej
Dla populacji nieeuropejskich dziś dostępne gotowe genetyczne wskaźniki ryzyka mogą działać znacznie gorzej, potencjalnie pogłębiając nierówności zdrowotne. Ta praca pokazuje, że uczenie transferowe — inteligentne dopracowanie istniejących wskaźników opartych na danych europejskich przy użyciu umiarkowanych zestawów danych z grup niedoreprezentowanych — może zamknąć znaczną część tej luki. W praktyce oznacza to, że systemy opieki zdrowotnej i badacze mogą budować bardziej dokładne i sprawiedliwe narzędzia genetyczne bez łączenia surowych danych między instytucjami czy pochodzeniami, co łagodzi obawy prywatności. Choć żadna pojedyncza metoda nie będzie najlepsza dla każdej cechy i populacji, zestaw narzędzi GPTL pokazuje, że sprawiedliwsza predykcja genetyczna jest technicznie osiągalna, jeśli potraktujemy przeszłe modele nie jako produkty ostateczne, lecz jako punkty wyjścia, które można dopasować do wszystkich."}
Cytowanie: Wu, H., Pérez-Rodríguez, P., Boehnke, M. et al. Improving polygenic score prediction for underrepresented groups through transfer learning. Nat Commun 17, 1973 (2026). https://doi.org/10.1038/s41467-026-68696-7
Słowa kluczowe: poligeniczne wskaźniki ryzyka, uczenie transferowe, predykcja genetyczna, nierówności zdrowotne, genetyka populacji