Clear Sky Science · pl
ProteoAutoNet: wysokoprzepustowa analiza współelucyjnych białek z użyciem robotyki i uczenia maszynowego
Dlaczego zrozumienie partnerstw białkowych ma znaczenie
W każdej komórce białka rzadko działają samotnie. Tworzą zmienne sojusze, aby budować struktury, kopiować DNA, usuwać uszkodzone elementy i napędzać wzrost. Wiele nowotworów przejmuje te partnerstwa, ale ich szczegółowe odwzorowanie postępowało wolno i żmudnie. W tym badaniu wprowadzono ProteoAutoNet, system wspierany robotyką i uczeniem maszynowym, który znacznie przyspiesza wykrywanie partnerstw białkowych w komórkach i pokazuje, jak podejście to może ujawnić ukryte słabe punkty w rakach tarczycy.
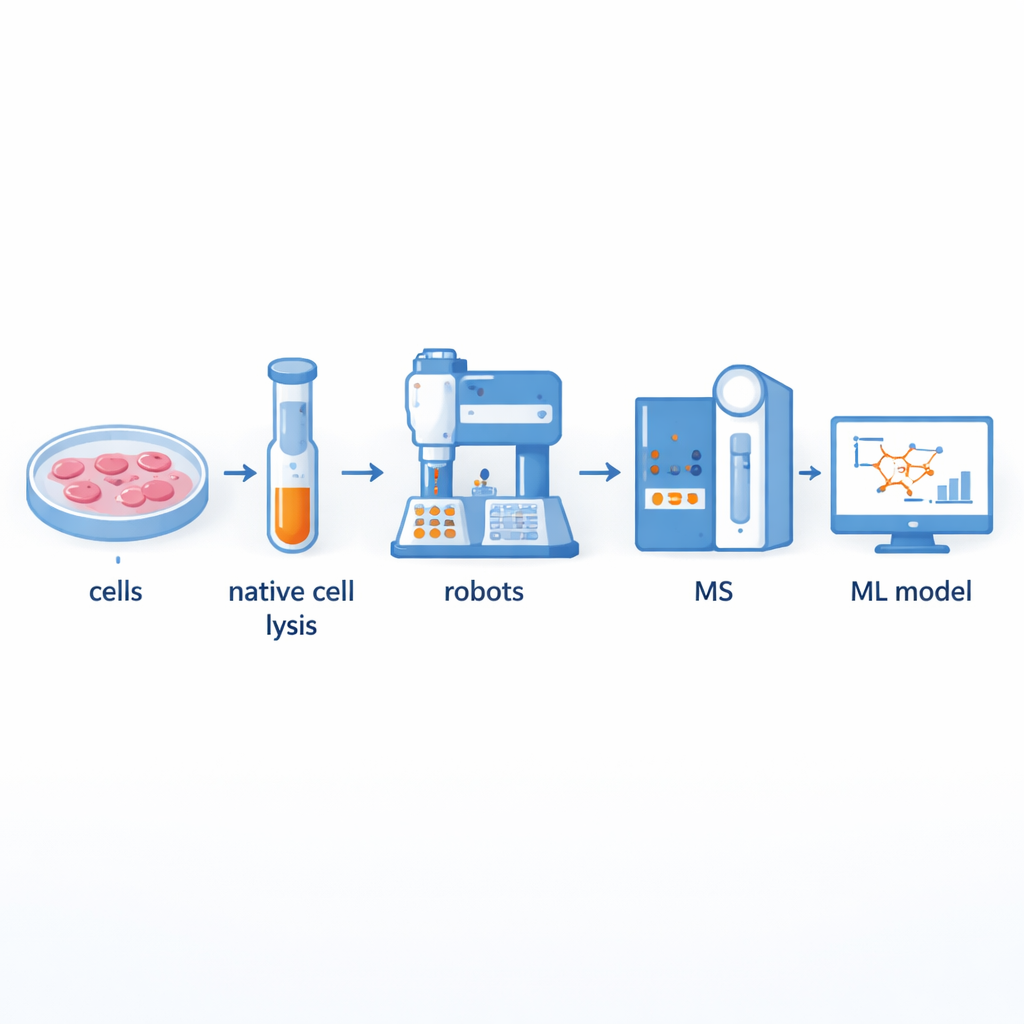
Budowa szybszej fabryki partnerstw białkowych
Tradycyjnie naukowcy stosują metodę zwaną ko-frakcjonowaniem z użyciem spektrometrii mas, aby rozdzielić duże kompleksy białkowe, a następnie zidentyfikować ich składniki. Chociaż potężna, ta metoda jest pracochłonna i ma niską przepustowość: ręczne przygotowanie setek frakcji może zająć wiele dni. Autorzy zbudowali platformę wspomaganą robotyką, która automatyzuje większość tego procesu. Zawartość komórek jest najpierw delikatnie uwalniana, tak by naturalne kompleksy białkowe pozostały nietknięte, potem przepuszczana przez kolumny rozdzielające według wielkości na dziesiątki frakcji. Roboty do manipulacji cieczami i ramiona robotyczne przejmują potem zadania, dodając odczynniki, trawiąc białka na mniejsze fragmenty, oczyszczając próbki i dostarczając je do spektrometru mas do pomiaru. Takie ustawienie może przetworzyć do 540 frakcji z wielu linii komórek tarczycy w ciągu zaledwie dwóch–trzech dni, co mniej więcej podwaja przepustowość w porównaniu z wcześniejszymi systemami półautomatycznymi.
Roboty, które są nie tylko szybsze, lecz także bardziej niezawodne
Samo tempo to za mało, jeśli wyniki są hałaśliwe lub niespójne. Zespół dokładnie sprawdził, czy automatyczny pipeline dorównuje lub przewyższa jakość tradycyjnego przetwarzania ręcznego. Używając próbek kontrolnych jakości, wykazali, że zautomatyzowany system wielokrotnie identyfikował niemal 3 000 białek na linię komórek tarczycy z bardzo dużym nakładaniem się między replikatami i silną zgodnością w zmierzonych ilościach białek. Gdy porównali bezpośrednio przetwarzanie robotyczne i ręczne tych samych próbek, obie metody wykrywały podobną liczbę białek, ale metoda robotyczna dawała nieco mniejszą zmienność w liczbach i bardziej stabilne pomiary obfitości białek. Oznacza to, że nowa platforma nie tylko oszczędza czas i pracę, lecz także wspiera bardziej odtwarzalne eksperymenty — co jest kluczowe przy dużych badaniach i zastosowaniach klinicznych.
Nauczanie komputerów rozpoznawania istotnych powiązań
Nawet przy szybkich urządzeniach pozostaje zasadnicze wyzwanie: jak zdecydować, które białka rzeczywiście wchodzą w interakcje, a które pojawiają się razem przypadkowo. Aby temu sprostać, autorzy połączyli skuratorowane bazy danych kompleksów białkowych z modelem uczenia maszynowego opartym na algorytmie XGBoost. Najpierw oczyścili i scalili trzy główne zasoby dotyczące kompleksów białkowych, kończąc z 96 635 znanymi interakcjami białko–białko. Następnie użyli profili pojawiania się białek w frakcjach jako cech wejściowych i oznaczyli pary jako prawdopodobnych partnerów lub nie-partnerów na podstawie baz danych. Ponieważ prawdziwe, wysokozaufane partnerstwa są stosunkowo rzadkie, zastosowali ukierunkowaną strategię augmentacji danych: stworzyli wiele nieznacznie zmodyfikowanych wersji znanych pozytywnych przykładów, aby nauczyć model rozpoznawania trwałych wzorców, a nie zapamiętywania konkretnych śladów. Trenowany na dziesiątkach milionów takich przykładów z trzech linii komórek tarczycy, model osiągnął dobrą wydajność, poprawnie klasyfikując prawdziwe interakcje znacznie powyżej poziomu losowego zarówno w testach wewnętrznych, jak i na niezależnej linii walidacyjnej.
Nowe spojrzenia na maszynerię komórek nowotworowych
Uzbrojeni w ten workflow, badacze zmapowali sieci interakcji w normalnej linii komórek tarczycy oraz w dwóch liniach nowotworowych: linii raka brodawkowatego tarczycy i linii raka pęcherzykowego, która potrafi dawać przerzuty do płuc. W tych komórkach zidentyfikowali ponad 25 000 prawdopodobnych interakcji białkowych i znaleźli silne sygnały od dobrze znanych „maszyn” komórkowych, takich jak rybosomy (które budują białka) i proteasomy (które je rozkładają), potwierdzając, że metoda odtwarza ustaloną biologię. Porównując komórki nowotworowe z linią normalną, odkryli sieci, które były w chorobie wzmocnione. W komórkach raka pęcherzykowego dającego przerzuty składniki proteasomu oraz kompleks opiekuńczy zwany prefoldyną były wyraźnie bardziej połączone i obfitsze. Kilka podjednostek prefoldyny wcześniej wiązano z innymi nowotworami, ale globalne badania białek przegapiły ich skoordynowane zachowanie w raku tarczycy — być może dlatego, że te białka są ściśle kontrolowane przez degradację. Podejście ko-frakcjonowania ujawniło ich skoordynowane zmiany na poziomie kompleksów.
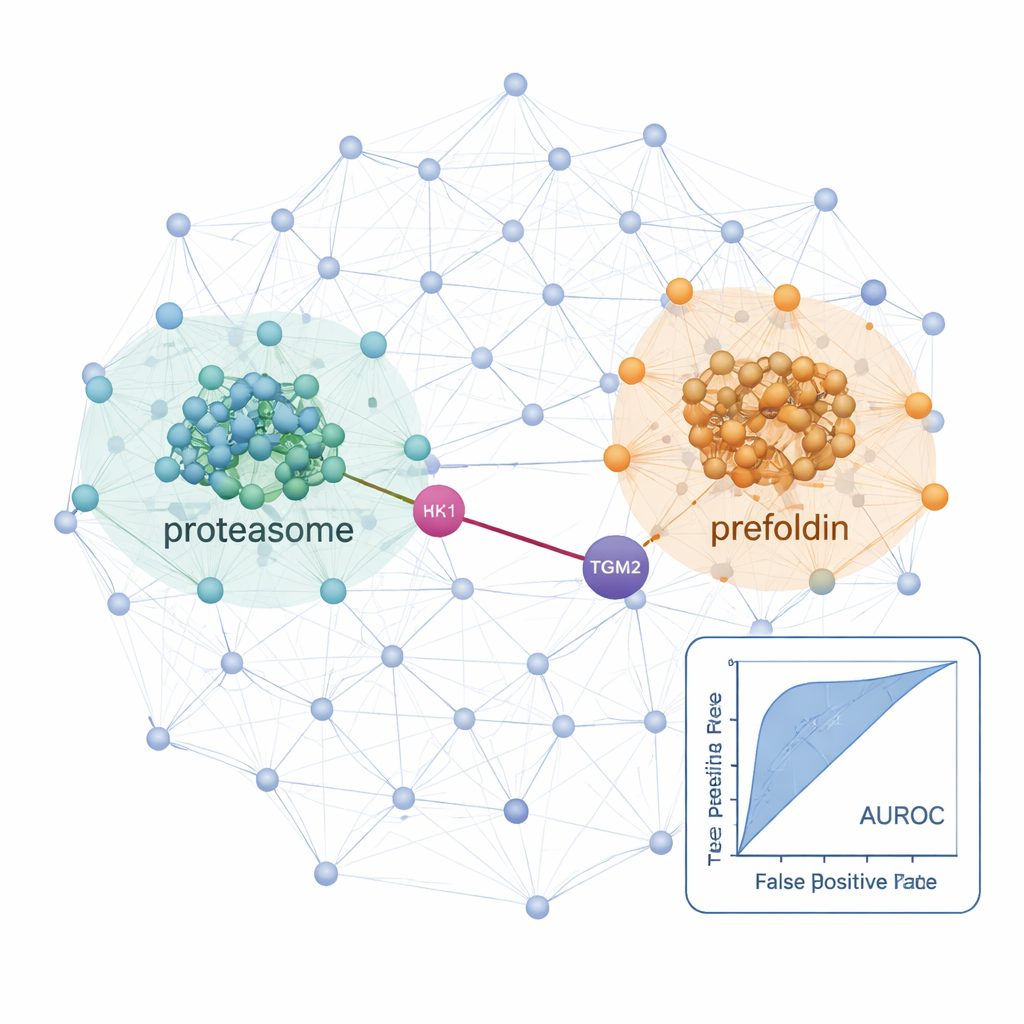
Ukryte powiązania, które mogą nakierować przyszłe terapie
Badanie wyróżniło także konkretne interakcje, które mogą mieć znaczenie dla tego, jak raki tarczycy rosną i dają przerzuty. Jednym przykładem jest przewidywane partnerstwo między HK1, enzymem inicjującym główną ścieżkę spalania cukrów w komórce, a TGM2, białkiem znanym z pobudzania inwazji i przerzutów w guzach tarczycy. To połączenie HK1–TGM2, nieobecne w istniejących bazach interakcji, zostało wsparte modelowaniem strukturalnym i wydawało się szczególnie aktywne w linii raka brodawkowatego, sugerując, że przeprogramowanie metaboliczne i zachowania inwazyjne mogą być ze sobą fizycznie powiązane. Podsumowując, ProteoAutoNet pokazuje, jak połączenie robotyki i uczenia maszynowego może przekształcić powolne, wymagające ekspertów mapowanie sieci białkowych w bardziej skalowalny proces. Dla czytelników niebędących specjalistami kluczowy przekaz jest taki, że ta technologia może ujawnić zarówno szerokie przesunięcia w maszynerii komórkowej, jak i nieoczekiwane partnerstwa białkowe, które mogą w przyszłości pomóc lekarzom lepiej przewidywać, które raki tarczycy będą zachowywać się agresywnie, oraz sugerować nowe cele terapii.
Cytowanie: Lyu, M., Hu, P., Zhang, G. et al. ProteoAutoNet: high-throughput co-eluted protein analysis with robotics and machine learning. Nat Commun 17, 1949 (2026). https://doi.org/10.1038/s41467-026-68686-9
Słowa kluczowe: interakcje białek, spektrometria mas, uczenie maszynowe w biologii, rak tarczycy, proteasom i prefoldyna