Clear Sky Science · pl
Kompleksowe porównanie metod tworzenia poligenicznych ocen ryzyka dla pojedynczych i wielo‑pochodzeniowych populacji za pomocą platformy PGS-hub
Dlaczego Twój wynik ryzyka na podstawie DNA ma znaczenie
Lekarze coraz lepiej odczytują nasze DNA, by oszacować, kto jest bardziej narażony na rozwój powszechnych chorób, takich jak choroby serca, cukrzyca czy schizofrenia. Takie oszacowania, zwane ocenami poligenicznymi, łączą maleńkie efekty wielu wariantów genetycznych w jedną liczbę. Obecnie istnieje wiele konkurencyjnych metod ich obliczania i nie działają one jednakowo dobrze dla osób o różnych pochodzeniach. W tym badaniu autorzy porównali najważniejsze metody bezpośrednio ze sobą i stworzyli internetową usługę, PGS-hub, która pozwala badaczom obliczać te oceny w sposób spójny i prosty.
Jedno miejsce do obliczania ocen ryzyka z DNA
Autorzy stworzyli PGS-hub — platformę internetową, która ukrywa większość technicznej złożoności związaną z ocenami poligenicznymi. Użytkownicy przesyłają wyniki badań genetycznych podsumowujące, jak miliony markerów DNA wiążą się z daną chorobą lub cechą. Następnie wybierają pochodzenie populacji, które ich interesuje — na przykład europejskie lub afrykańskie — i wybierają z menu jedną z popularnych metod obliczeń. W tle PGS-hub konwertuje dane wejściowe do właściwych formatów, korzysta z gotowych paneli referencyjnych opisujących korelacje między pobliskimi markerami DNA i uruchamia dużą liczbę zadań na systemie wysoko wydajnych obliczeń. Wynikiem jest skompresowany plik wag, który można zastosować do genotypów poszczególnych osób, by wygenerować ich wynik. 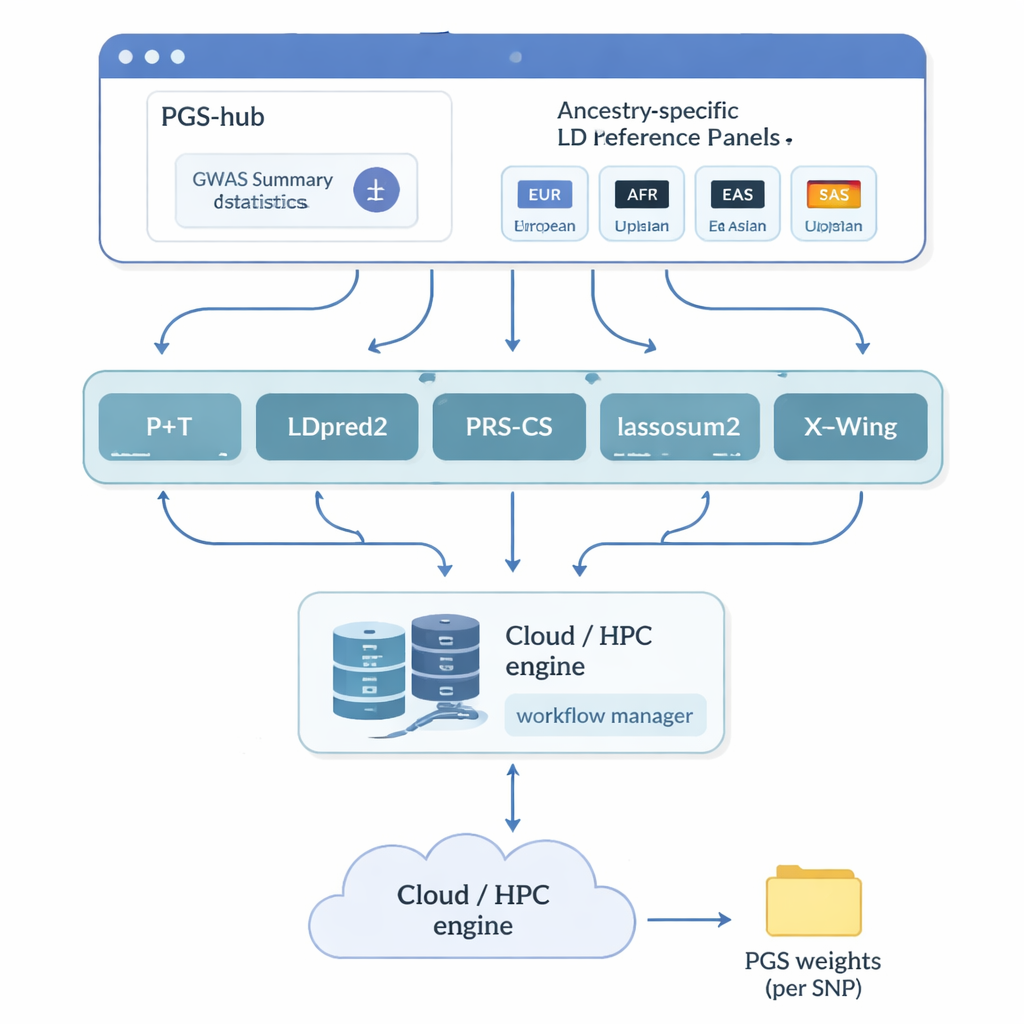
Test 13 metod obliczania
Aby sprawdzić, które podejścia działają najlepiej, zespół porównał 13 najnowocześniejszych metod w 36 chorobach i cechach u niemal 380 000 osób pochodzenia europejskiego oraz nieco ponad 8 000 osób pochodzenia afrykańskiego z UK Biobank. Oceniano nie tylko, jak dobrze każda ocena przewidywała obecność choroby lub wyższe wartości cechy, lecz także ile czasu obliczeniowego i pamięci wymagała każda metoda. W populacji europejskiej jedna metoda o nazwie LDpred2 zazwyczaj dostarczała najbardziej dokładne wyniki, często przewyższając inne metody wyraźnie. Kilka alternatyw — lassosum2, PRS-CS i SDPR — w wielu cechach radziło sobie niemal tak dobrze, podczas gdy starsze metody pozostawały w tyle. Dla cech takich jak wzrost czy choroba Leśniowskiego‑Crohna najlepsze oceny wyjaśniały znaczną część ryzyka genetycznego; dla innych, na przykład funkcji nerek, wszystkie metody miały trudności, co odzwierciedla słabsze sygnały genetyczne leżące u podstaw tych cech.
Wnioski dla zróżnicowanych populacji i metod łączonych
Głównym problemem w predykcji genetycznej jest to, że metody trenowane głównie na Europejczykach mogą nie przenosić się dobrze na osoby o innym pochodzeniu. Kiedy autorzy powtórzyli benchmarking z użyciem badań genetycznych osób pochodzenia afrykańskiego, każda metoda działała gorzej, podkreślając brak dużych badań w tych populacjach. Mimo to LDpred2 i SDPR zwykle należały do lepszych opcji. Zespół zbadał także podejścia „wielo‑pochodzeniowe”, które jawnie łączą informacje z różnych populacji. Tutaj stosunkowo prosta strategia — liniowe łączenie najlepszych wyników LDpred2 dla poszczególnych pochodzeń w pojedynczy wynik LDpred2-multi — przewyższyła bardziej złożone modele wielo‑pochodzeniowe, takie jak PRS-CSx czy X-Wing, zarówno dla grup europejskich, jak i afrykańskich. Dodatkowo autorzy pokazali, że zbudowanie zespołu (ensemble), który miesza najsilniejsze wyniki z różnych metod, dodatkowo poprawia predykcję dla wszystkich cech, szczególnie dla wysoce dziedzicznych chorób, takich jak schizofrenia czy choroba wieńcowa. 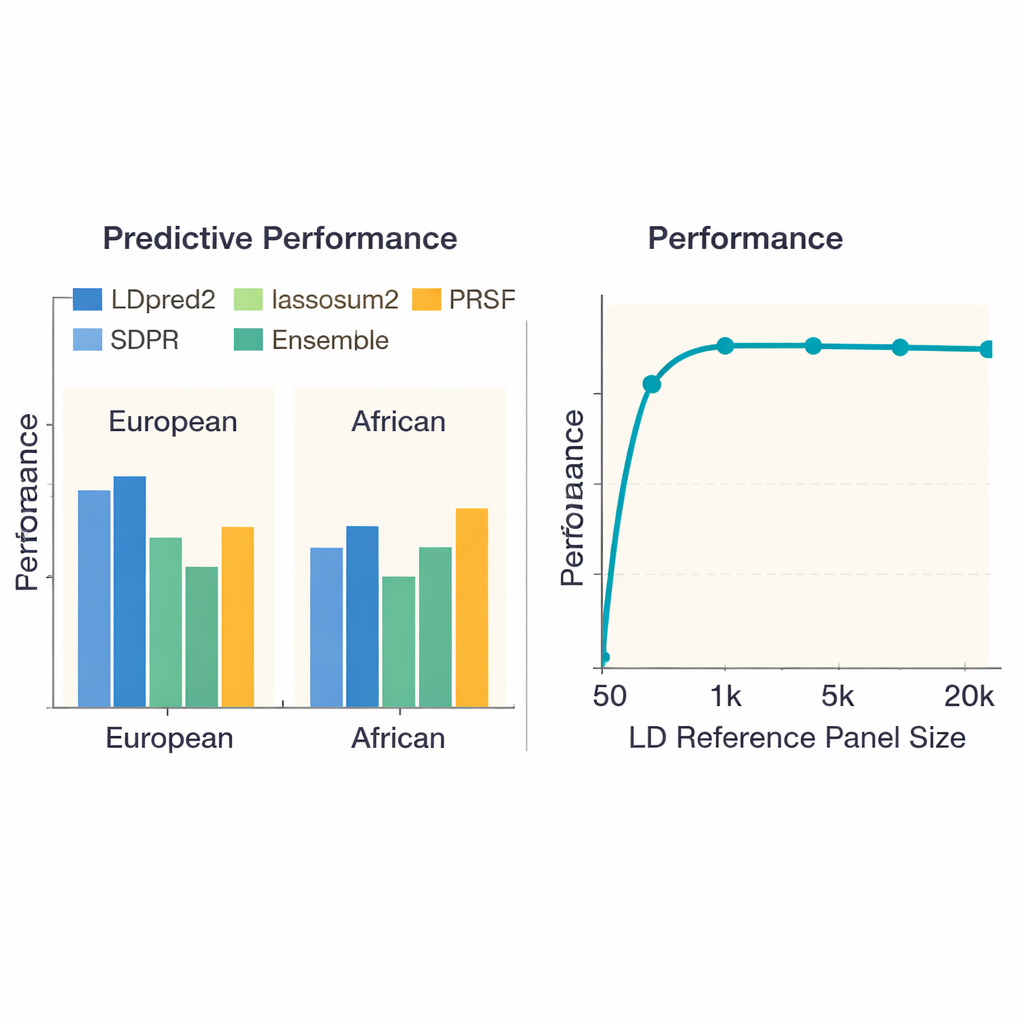
Jak wybory danych i ograniczenia obliczeniowe wpływają na oceny
Badanie sprawdziło, jak rozmiar panelu referencyjnego — zbioru osób używanych do poznania współzmienności pobliskich markerów DNA — wpływa na wydajność. Gdy panel był bardzo mały (mniej niż 1 000 osób), oceny były wyraźnie mniej dokładne. W miarę zwiększania panelu do około 5 000 osób wydajność poprawiała się gwałtownie, a następnie ustabilizowała, co sugeruje, że coraz większe panele przynoszą malejące korzyści. Co zaskakujące, samo dodanie większej liczby markerów DNA nie zawsze pomagało: użycie około 6,6 miliona wariantów czasami pogarszało przewidywania w porównaniu z ostro dobranym zbiorem około 1,1 miliona, prawdopodobnie dlatego, że dodatkowe markery wprowadzały więcej szumu niż użytecznego sygnału. Autorzy udokumentowali też duże różnice w kosztach obliczeniowych. Proste metody, takie jak podstawowe przycinanie i progowanie, kończyły pracę w mniej niż godzinę na cechę, podczas gdy niektóre podejścia bayesowskie wymagały setek godzin CPU — informacja istotna dla dużych projektów lub grup o ograniczonych zasobach.
Co to oznacza dla przyszłej predykcji na podstawie DNA
Dla osób niebędących specjalistami kluczowy przekaz jest taki, że nie wszystkie oceny ryzyka oparte na DNA są jednakowe, a szczegóły ich budowy mocno wpływają na to, kto może z nich skorzystać. Ta praca daje praktyczne wskazówki: metody takie jak LDpred2 i dobrze zaprojektowane zespoły zazwyczaj dają najbardziej wiarygodne przewidywania w dużych europejskich zbiorach danych, a kombinacje wielo‑pochodzeniowe mogą przewyższać bardziej skomplikowane modele między‑populacyjne. Jednocześnie spadek dokładności u osób pochodzenia afrykańskiego podkreśla pilną potrzebę większych i bardziej zróżnicowanych badań genetycznych. Poprzez zebranie wielu metod w jednej, ustandaryzowanej platformie online, PGS-hub obniża barierę dla badaczy na całym świecie, umożliwiając generowanie i porównywanie ocen poligenicznych — ważny krok w kierunku sprawiedliwego i skutecznego wykorzystania tych ocen w medycynie.
Cytowanie: Chen, X., Wang, F., Zhao, H. et al. Comprehensive benchmarking single and multi ancestry polygenic score methods with the PGS-hub platform. Nat Commun 17, 2014 (2026). https://doi.org/10.1038/s41467-026-68599-7
Słowa kluczowe: oceny poligeniczne, predykcja ryzyka genetycznego, platforma PGS-hub, genomika wielo‑pochodzeniowa, UK Biobank