Clear Sky Science · pl
TET1 jako główny regulator kontrolujący nadzór ferroptozy zależnej i niezależnej od GPX4 w ostrej białaczce szpikowej
Dlaczego te badania mają znaczenie dla leczenia nowotworów
Wiele nowych leków przeciwnowotworowych ma na celu sprowokowanie komórek nowotworowych do autodestrukcji w postaci ferroptozy — typu śmierci komórkowej napędzanej przez żelazo i uszkodzenia lipidów. Niektóre guzy jednak opierają się temu mechanizmowi. Niniejsze badanie ujawnia, jak białko modyfikujące DNA o nazwie TET1 pomaga komórkom białaczkowym unikać ferroptozy przez dwa odrębne systemy biochemicznej obrony — i pokazuje, że blokowanie tych mechanizmów może uczynić nawet oporne nowotwory podatnymi na terapię.
Śmiertelna mieszanka żelaza i uszkodzonych tłuszczów
Ferroptoza zachodzi, gdy żelazo napędza niekontrolowaną oksydację lipidów w błonach komórkowych, co ostatecznie prowadzi do pęknięcia komórek. W ostrej białaczce szpikowej (AML), podobnie jak w wielu nowotworach, komórki uruchamiają potężne systemy nadzoru, by powstrzymać ten proces. Jednym z kluczowych strażników jest enzym GPX4, który wykorzystuje małą cząsteczkę zwaną glutationem do neutralizacji szkodliwych nadtlenków lipidowych. Inne systemy zapasowe wytwarzają przeciwutleniacze, które mogą wychwytywać niebezpieczne rodniki nawet gdy GPX4 jest niesprawny. Zrozumienie, które „główne włączniki” koordynują te mechanizmy obronne, jest kluczowe dla zaprojektowania terapii, które niezawodnie wywołają ferroptozę w komórkach nowotworowych, oszczędzając tkanki zdrowe.
TET1 wyłania się jako centralny węzeł kontrolny
Naukowcy porównali dziesiątki próbek komórek nowotworowych, w tym wiele linii AML i komórek pochodzących od pacjentów, i zauważyli wyraźny wzorzec: komórki oporne na ferroptozę miały wyższe poziomy TET1, enzymu zmieniającego chemiczne znaczniki DNA i wpływającego na aktywność genów. Gdy obniżono poziom TET1 przy użyciu narzędzi genetycznych lub zahamowano jego aktywność małą cząsteczką, komórki nowotworowe stały się wyraźnie bardziej wrażliwe na leki wywołujące ferroptozę. Obserwacje te potwierdziły się zarówno w warunkach laboratoryjnych, jak i w modelach mysich AML. Odwrotnie, zwiększenie ekspresji TET1 chroniło komórki przed śmiercią ferroptotyczną i ograniczało gromadzenie reaktywnych form tlenu — chemicznie agresywnych produktów ubocznych, które napędzają uszkodzenie błon.
Wzmacnianie głównej tarczy antyoksydacyjnej
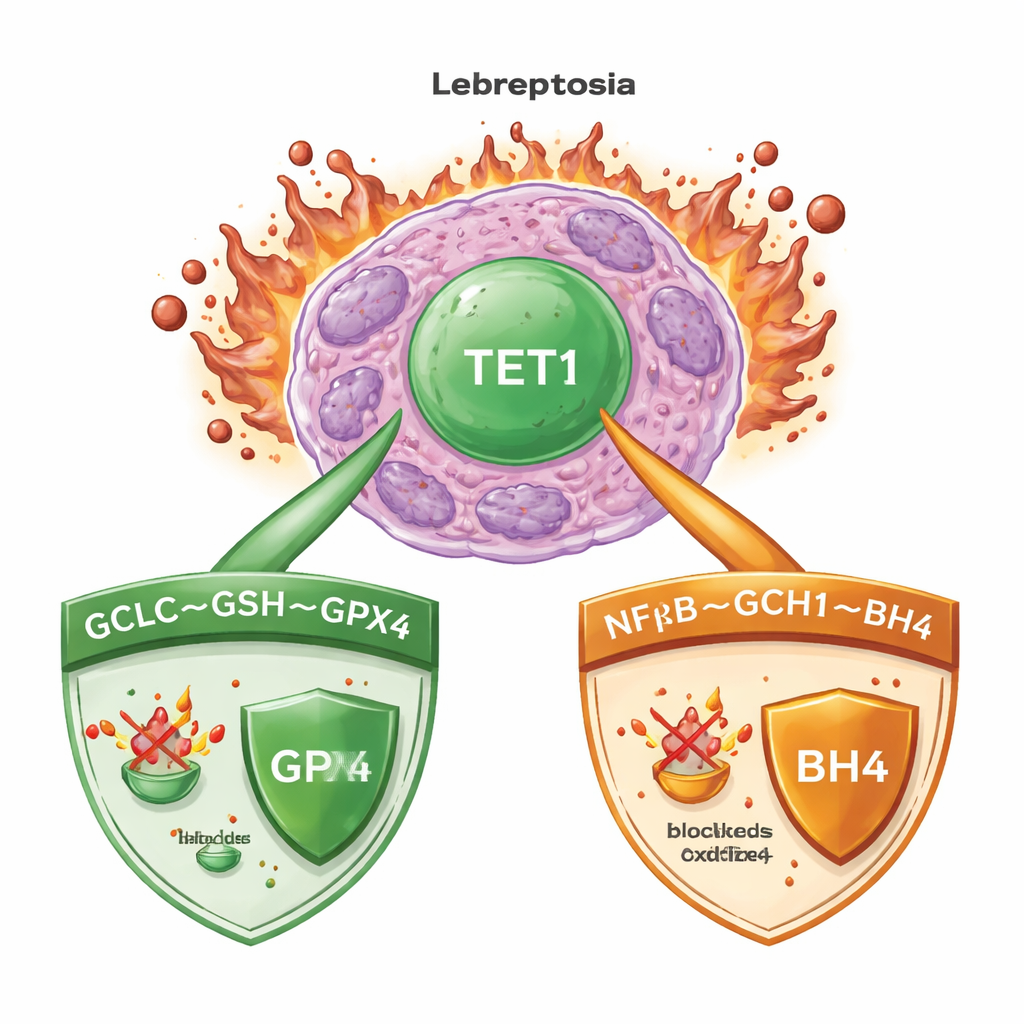
Zagłębiając się dalej, zespół zmapował miejsca działania TET1 w genomie i odkrył, że bezpośrednio aktywuje gen o nazwie GCLC. GCLC koduje kluczowy enzym inicjujący produkcję glutationu — paliwa dla GPX4. Poprzez zwiększanie specyficznego znacznika DNA (5-hydroksymetylocytozyny) w regionie promotora GCLC, TET1 zwiększa syntezę glutationu. W normalnych warunkach odżywczych to podnosi główną pulę antyoksydantów komórki. Podczas niedoboru cystyny ten sam kompleks enzymatyczny wytwarza nietypowe γ-glutamylowe peptydy, które pomagają pochłaniać nadmiar glutaminianu — kolejny sposób tłumienia ferroptozy. Zarówno w hodowlach komórkowych, jak i u myszy, utrata TET1 lub farmakologiczne zahamowanie syntezy glutationu ostro obniżały poziomy glutationu i tych ochronnych peptydów, czyniąc komórki białaczkowe znacznie bardziej podatnymi na wyzwalacze ferroptozy.
Druga, niezależna od GPX4 droga ucieczki
Zaskakująco, rola ochronna TET1 nie kończyła się na osi glutation–GPX4. Nawet gdy GPX4 został usunięty z komórek białaczkowych, dodatkowy TET1 wciąż mógł zapobiec śmierci ferroptotycznej, co sugerowało istnienie drugiej linii obrony. Autorzy powiązali to z aktywacją przez TET1 szlaku sygnałowego NFκB, w szczególności składnika NFKB2. To z kolei zwiększa ekspresję GCH1, enzymu produkującego przeciwutleniacz BH4. BH4 może chronić lipidy błonowe przed oksydacją bez udziału GPX4. Gdy GCH1 został wyciszony genetycznie lub zablokowany chemicznie, zdolność TET1 do ochrony komórek przed ferroptozą została częściowo utracona. Razem wyniki te definiują drogę TET1–NFKB2–GCH1 jako system nadzoru ferroptozy niezależny od GPX4.
Przekształcenie słabości w okazję terapeutyczną
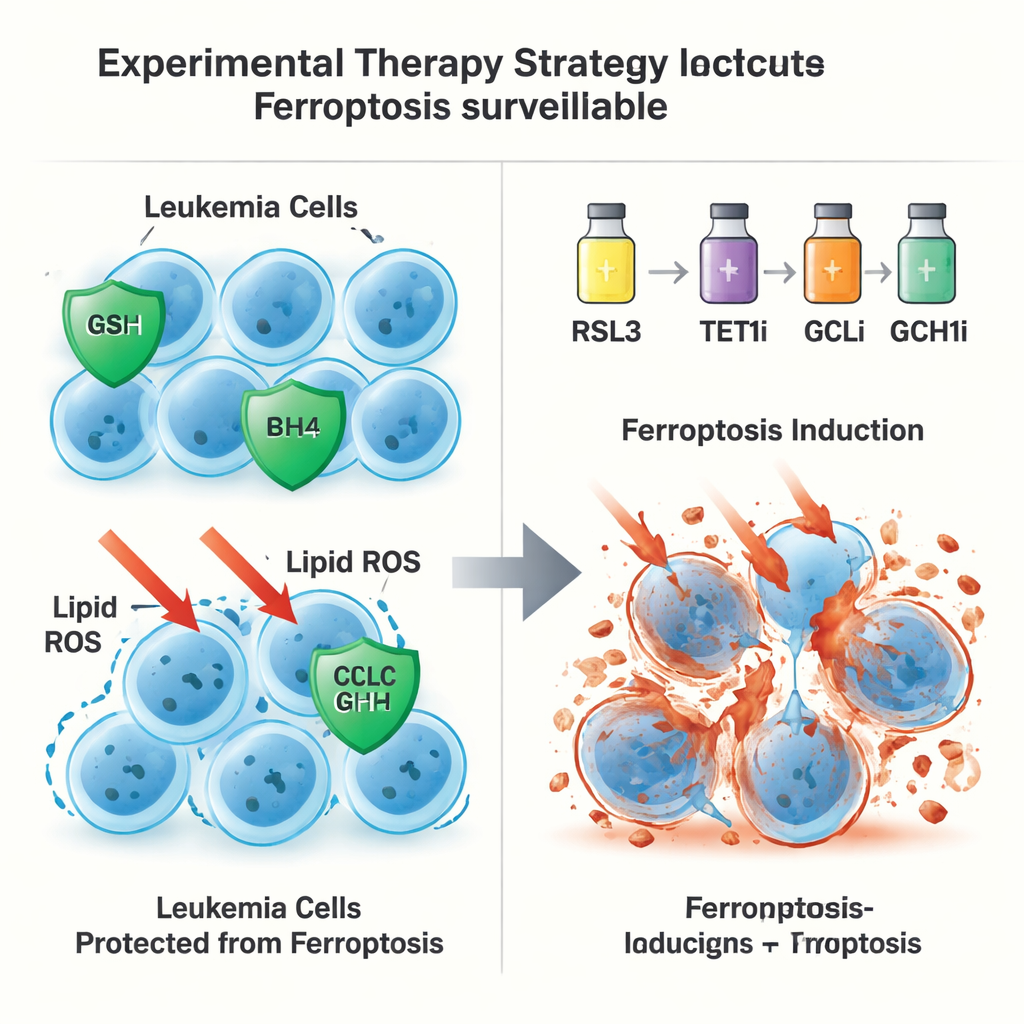
Uzbrojeni w tę mapę dwóch ścieżek, badacze sprawdzili, czy jednoczesne pobudzenie ferroptozy i osłabienie obron kontrolowanych przez TET1 może dać przewagę terapeutyczną. W modelach mysich AML oraz w przeszczepach białaczkowych pochodzących od pacjentów u myszy, niskie dawki leku wywołującego ferroptozę w połączeniu z inhibitorami TET1, syntezy GSH (przez GCLC) lub GCH1 dramatycznie zmniejszały obciążenie białaczką, wydłużały przeżycie i wyczerpywały populacje komórek inicjujących białaczkę. Co ważne, induktor ferroptozy był stosowany w ułamku dawek raportowanych w wcześniejszych badaniach, zmniejszając obawy o toksyczność wobec normalnych komórek macierzystych krwi.
Co to oznacza dla przyszłych terapii przeciwnowotworowych
Dla osób niebędących specjalistami kluczowa wiadomość jest taka, że komórki białaczkowe przetrwają dzięki dwóm nakładającym się „tarcza antyoksydacyjnym”, obydwu koordynowanym przez TET1: jednej skoncentrowanej na glutationie i GPX4, i drugiej opartej na GCH1 i BH4. Praca ta pokazuje, że przez umiarkowane pobudzenie ferroptozy przy jednoczesnym blokowaniu TET1 i jego dalszych partnerów, lekarze mogą w przyszłości pokonać oporność i selektywnie sprowadzić komórki nowotworowe do śmierci, nie przeciążając tkanek zdrowych. Chociaż te strategie nie są jeszcze gotowe do zastosowań klinicznych, badanie wskazuje TET1 jako potężny węzeł kontrolny i obiecujący cel do terapii skojarzonych w AML i potencjalnie w innych trudnych do leczenia nowotworach.
Cytowanie: Yang, L., Lu, J., Yun, W. et al. TET1 as a master regulator controlling GPX4-dependent and -independent ferroptosis surveillance in acute myeloid leukemia. Nat Commun 17, 1800 (2026). https://doi.org/10.1038/s41467-026-68509-x
Słowa kluczowe: ferroptoza, ostra białaczka szpikowa, TET1, glutation, epigenetyka raka