Raki płuc oporne na inhibitory EGFR wykazują nadwrażliwość krzyżową na kowalencyjny, niezależny od cysteiny mostkujący oligomeryzujący cząsteczkowy układ KEAP1
Dlaczego oporne na leki nowotwory płuc mają znaczenie
Leki ukierunkowane zrewolucjonizowały leczenie niektórych raków płuc, celując w wadliwy sygnał wzrostu zwany EGFR. Jednak u większości pacjentów te terapie przestają działać w ciągu kilku lat, gdy nowotwór rozwija oporność. Badanie to ujawnia zaskakujący zwrot akcji: gdy guzy stają się oporne na inhibitory EGFR, pojawia się nowe piętno Achillesa, które można zaatakować innym typem związku. Zrozumienie tej ukrytej słabości może zainspirować przyszłe strategie leczenia, które będą zamykać ewolucyjne ścieżki nowotworu zamiast ciągłego ich doganiania.
Ujawniona ukryta słabość
Naukowcy skupili się na niedrobnokomórkowych rakach płuc napędzanych mutacyjnym EGFR, powszechnej postaci choroby. W laboratorium porównali komórki nowotworowe wrażliwe na leki z blisko spokrewnionymi komórkami, które rozwinęły oporność na inhibitory blokujące EGFR, takie jak gefitynib i osimertynib. Następnie przetestowali bibliotekę około 2100 małych cząsteczek, aby sprawdzić, które z nich zabijają komórki oporne skuteczniej niż pierwotne, wrażliwe komórki. Spośród wielu kandydatów jedna substancja, nazwana MCB-613, wyróżniała się konsekwentnie. Komórki oporne na inhibitory EGFR, które je lekceważyły, okazały się wyjątkowo podatne na MCB-613, zarówno w hodowlach komórkowych, jak i w guzach mysich.
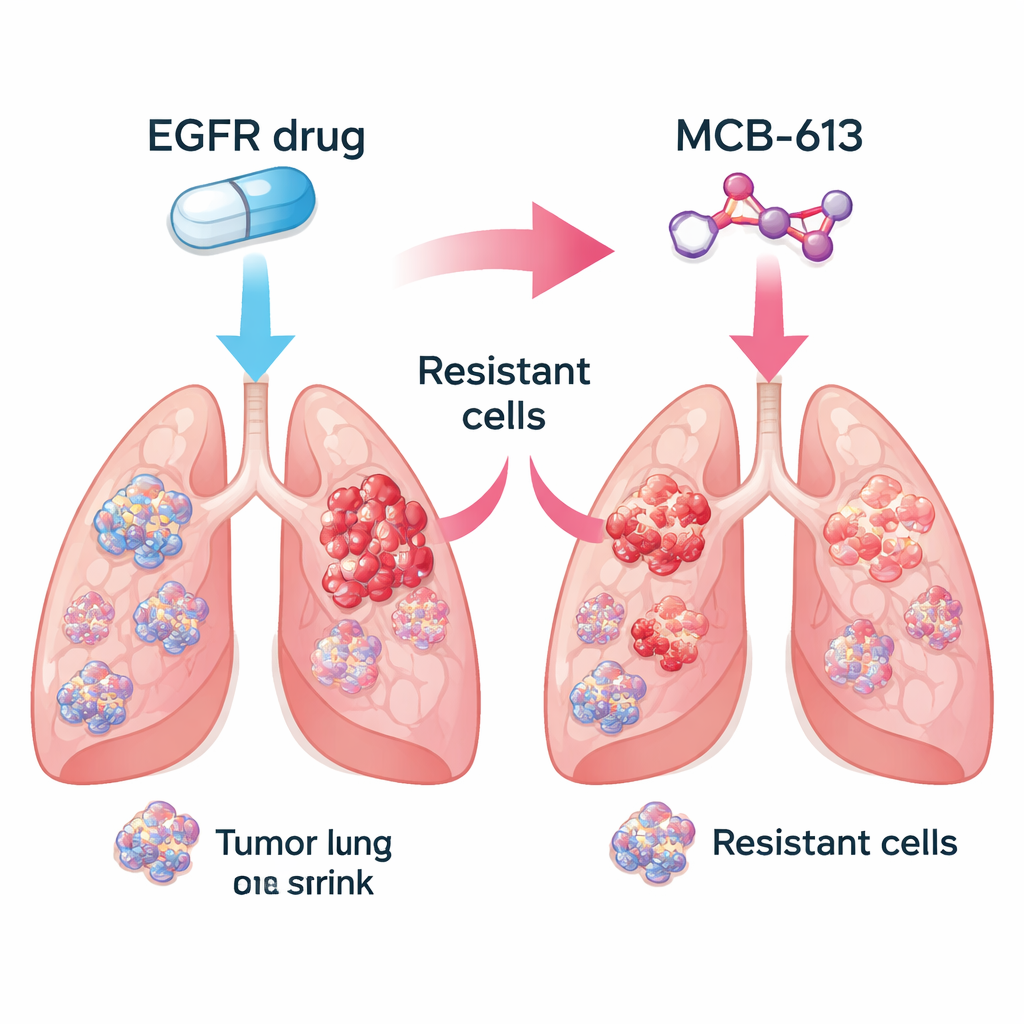
Uwięzienie zmieszanych populacji guza Figure 1.
Prawdziwe guzy to mieszaniny komórek: niektóre pozostają wrażliwe na pierwotny lek, podczas gdy inne zdobywają oporność poprzez różne genetyczne sztuczki. Zespół zapytał, czy połączenie inhibitora EGFR z MCB-613 mogłoby zlikwidować tę różnorodność. W kontrolowanym eksperymencie wymieszali głównie komórki wrażliwe na lek z niewielkim odsetkiem różnych typów komórek opornych, naśladując guz pacjenta. Leczenie tej zmieszanej populacji albo samym inhibitorem EGFR, albo tylko MCB-613 pozwalało pewnym komórkom przetrwać i rozwijać się. Jednak gdy zastosowano oba środki razem, cała populacja upadła. Sugeruje to, że połączenie standardowej terapii ukierunkowanej z dobrze dobranym lekiem wywołującym „nadwrażliwość krzyżową” może doprowadzić guzy do ewolucyjnego impasu.
Cząsteczkowy most, który łamie strażnika
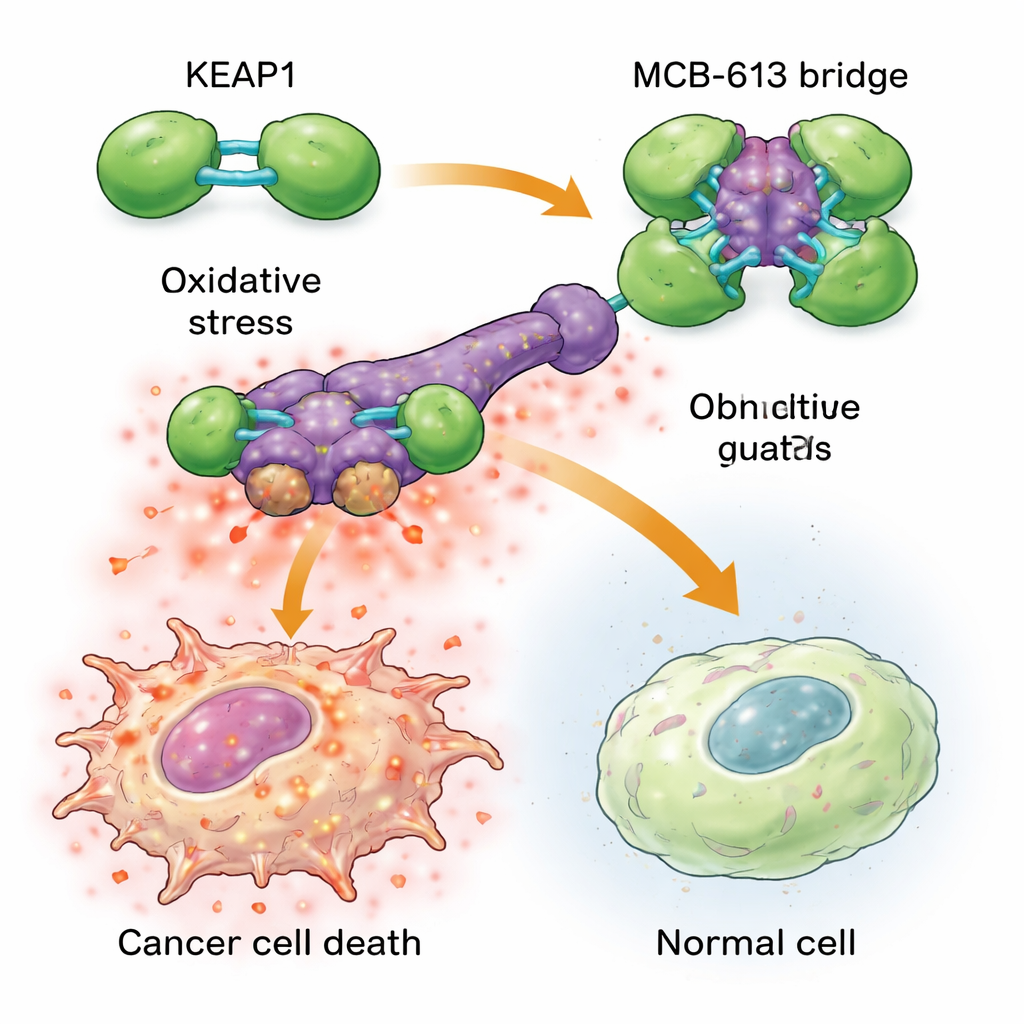
Aby zrozumieć, dlaczego MCB-613 tak mocno uderza w komórki oporne, naukowcy zbadali, do jakich białek się wiąże. Przy użyciu sond chemicznych i ukierunkowanego ekranu genowego CRISPR zidentyfikowali białko o nazwie KEAP1 jako niezbędne dla działania MCB-613. KEAP1 zwykle pełni rolę komórkowego strażnika, wyczuwając stres i pomagając regulować reakcje ochronne. Zespół odkrył, że MCB-613 przyłącza się do KEAP1 w nietypowy sposób: zachowuje się jak sztywny cząsteczkowy most, łącząc jednostki KEAP1 w nadmierne, nieprawidłowe skupiska. Proces ten nie zależy od zwykłych reaktywnych miejsc zawierających siarkę w KEAP1, lecz od specyficznego aminokwasu lizyny w regionie dymeryzacji. Gdy ta lizyna została zmutowana, MCB-613 nie mogło już sklejać KEAP1, a komórki oporne przestały być nadwrażliwe na związek.
Przekształcanie pomocnego stresu w śmiertelne przeciążenie Figure 2.
Zlepianie KEAP1 uruchamia niebezpieczną kaskadę w komórkach nowotworowych opornych na leki. Te komórki już żyją w podwyższonym tzw. stresie podstawowym, z podwyższonym poziomem reaktywnych form tlenu (szkodliwych produktów ubocznych) oraz zwiększoną aktywnością ochronnej sieci sygnalizacyjnej znanej jako zintegrowana odpowiedź na stres. Gdy dodano MCB-613, zaburzenie KEAP1 przepchnęło ten obciążony stan za próg: reaktywne formy tlenu jeszcze się nagromadziły, a kluczowe regulatory stresu, zwane ATF4 i CHOP, włączyły potężne programy śmierci. Blokowanie tych regulatorów stresu lub chemiczne usuwanie reaktywnych form tlenu w dużej mierze chroniło komórki przed MCB-613. Co ciekawe, klasyczny partner KEAP1, NRF2, często uważany za główny kierowcę obrony antyoksydacyjnej, nie odpowiadał za zabijanie; w rzeczywistości usunięcie NRF2 uczyniło komórki jeszcze bardziej wrażliwymi, co podkreśla, że MCB-613 wykorzystuje inną, niekanoniczną ścieżkę.
Co to może oznaczać dla przyszłych terapii
Na razie MCB-613 jest związkiem narzędziowym z chemicznymi wadami, które czynią go nieodpowiednim jako lek. Ale ujawnia potężną koncepcję: gdy raki płuc rozwijają oporność na inhibitory EGFR, mogą zostać uwięzione w stanie przewlekłego stresu, który można selektywnie zaatakować związkami zmuszającymi KEAP1 do tworzenia dysfunkcyjnych zespołów. W zasadzie dopracowane wersje takich „cząsteczkowych mostów” mogłyby zostać opracowane tak, by były bezpieczniejsze i bardziej precyzyjne, dając onkologom sposób na skierowanie guzów w „niemożliwy wybór” między wrażliwością na pierwotną terapię ukierunkowaną a wrażliwością na następną terapię wywołującą stres. Taka strategia ewolucyjnego uwięzienia mogłaby ostatecznie pomóc opóźnić lub przezwyciężyć oporność w raku płuc z mutacją EGFR, a być może także w innych trudno leczonych nowotworach.
Cytowanie: Bassil, C.F., Dillon, K., Anderson, G.R. et al. EGFR inhibitor-resistant lung cancers exhibit collateral sensitivity to a covalent, cysteine-independent KEAP1 oligomerizing molecular bridge.
Nat Commun17, 1726 (2026). https://doi.org/10.1038/s41467-026-68424-1
Słowa kluczowe: rak płuc z mutacją EGFR, oporność na leki, nadwrażliwość krzyżowa, KEAP1, stres oksydacyjny