Clear Sky Science · pl
Tmem110 reguluje konformację TRPML1, aby utrzymać homeostazę endolizosomalną i zapobiegać wydostawaniu się mitochondrialnego DNA oraz patologicznej obróbce własnego DNA
Dlaczego wyciek DNA po urazie ma znaczenie
Gdy doznajemy ciężkiego urazu, na przykład silnego uderzenia w głowę, szkoda nie kończy się w miejscu urazu. Wielu pacjentów rozwija groźną reakcję ogólnoustrojową, w której płuca, wątroba, nerki i inne narządy zaczynają zawodzić. Badanie to ujawnia, jak maleńkie struktury wewnątrz komórek — lizosomy, mitochondria i czujniki odpornościowe — komunikują się, by zdecydować, czy organizm spokojnie posprząta pozostałości, czy wpadnie w autoagresywną reakcję immunologiczną. Zrozumienie tego ukrytego systemu sprzątania może otworzyć nowe możliwości zapobiegania niewydolności narządów po urazie oraz leczenia chorób autoimmunologicznych i zapalnych.
Od urazu głowy do kryzysu ogólnoustrojowego
Urazowe uszkodzenie mózgu (TBI) może wywołać potężną burzę zapalną zwaną zespołem dysfunkcji wielonarządowej (MODS), jednak mechanizm rozprzestrzeniania się szkody z mózgu na odległe narządy pozostawał niejasny. Autorzy skupili się na monocytach i makrofagach — komórkach odpornościowych patrolujących krew i tkanki, działających jak zawodowi zbieracze odpadów. Po TBI komórki umierają w sposób niekontrolowany i uwalniają całe mitochondria — maleńkie fabryki energii posiadające własne DNA — do krążenia. To mitochondrialne DNA (mtDNA) wygląda dla układu odpornościowego podejrzanie podobnie do bakteryjnego DNA i może włączyć silne obronne reakcje w stylu przeciwwirusowym, jeśli nie zostanie szybko usunięte. Normalnie komórki internalizują te zabłąkane mitochondria do kwaśnych przedziałów zwanych lizosomami, gdzie enzymy rozkładają DNA zanim wycieknie do płynu komórkowego. 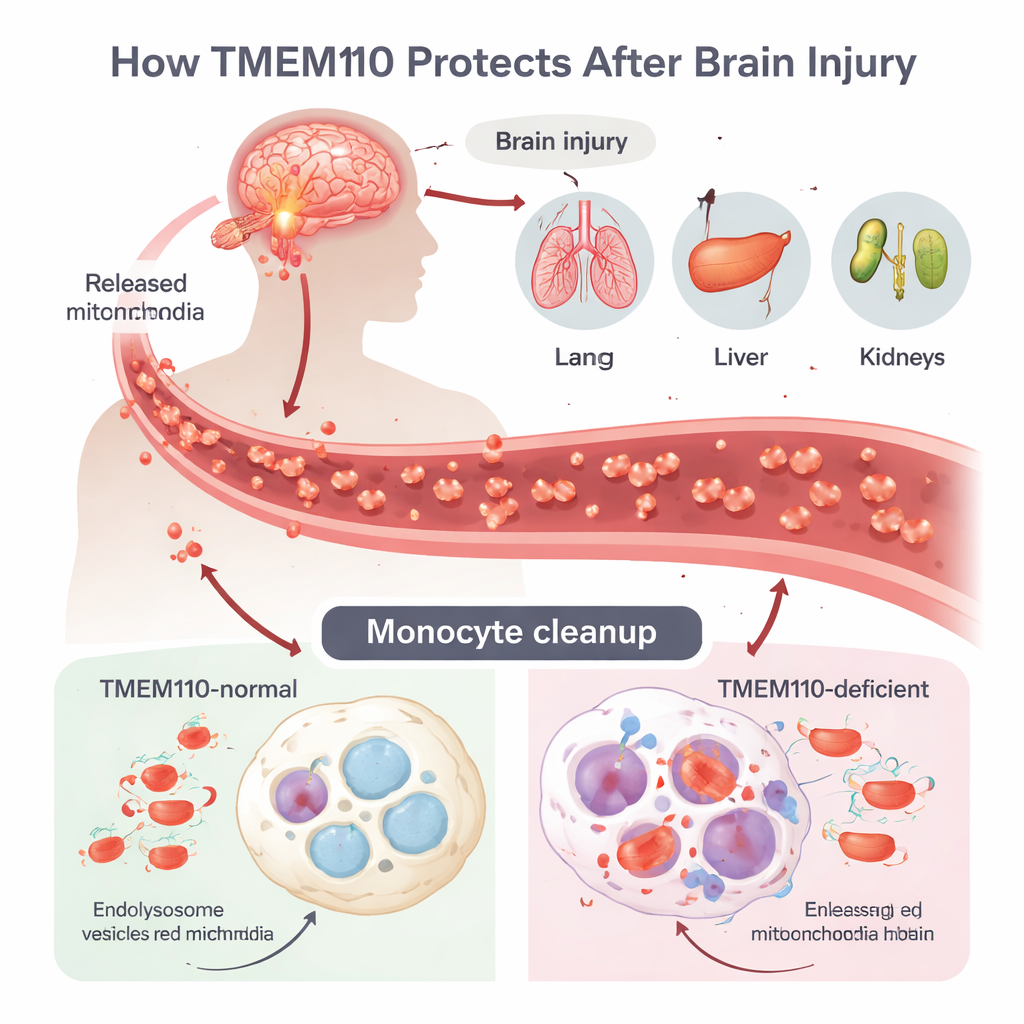
Bramkowy białko utrzymujące stabilność lizosomów
Zespół zidentyfikował TMEM110, białko przecinające błonę siateczki śródplazmatycznej (głównej sieci błon wewnątrzkomórkowych), jako kluczowego strażnika tego procesu. Używając genetycznie zmodyfikowanych myszy, u których TMEM110 był brakujący wyłącznie w monocytach i makrofagach, stworzyli kontrolowany model urazu mózgu. W porównaniu z normalnymi myszami, zwierzęta pozbawione TMEM110 rozwijały znacznie wyższe poziomy interferonów typu I — silnych cząsteczek sygnałowych przeciwwirusowych — we krwi i płynie mózgowo‑rdzeniowym. Ich płuca, wątroba, nerki i węzły chłonne wykazywały poważniejsze uszkodzenia, a długoterminowe przeżycie po TBI było znacząco gorsze. Kiedy badacze zablokowali receptor dla interferonów typu I lub zastąpili zmutowaną szpikę kostną normalnymi komórkami, uraz narządów i śmiertelność spadły, co pokazuje, że nadmierne sygnalizowanie interferonowe pochodzące z obwodowych monocytów jest kluczowym czynnikiem napędzającym MODS w tym kontekście.
Jak wadliwe gospodarowanie jonami pozwala DNA uciec
Wnikając do wnętrza tych komórek odpornościowych, autorzy stwierdzili, że lizosomy pozbawione TMEM110 były chemicznie niezrównoważone: miały zmniejszoną kwasowość i były przeciążone wapniem. W takich warunkach enzym DNase II — który normalnie sieka DNA w lizosomach — tracił aktywność, błony lizosomalne stawały się kruche, a mtDNA uciekało do otaczającej cytoplazmy. Eksperymenty laboratoryjne z ludzkimi monocytami pochłaniającymi oznakowane mitochondria dawcy potwierdziły, że bez TMEM110 więcej mtDNA przeciekało do płynu komórkowego i wywoływało gwałtowny wzrost genów stymulowanych interferonem. Badanie pokazuje, że TMEM110 działa przez kontrolowanie innego białka — lizosomalnego kanału jonowego TRPML1. Gdy TMEM110 jest obecny, nawiązuje fizyczny kontakt z TRPML1 i przestawia go w konformację „otwartą”, tworząc małe ogniska uwalniania wapnia na powierzchni lizosomu. Te kontrolowane wypływy wapnia pomagają utrzymać odpowiednią kwasowość, wspierają naprawę błony i utrzymują mtDNA uwięzione i degradowane wewnątrz. 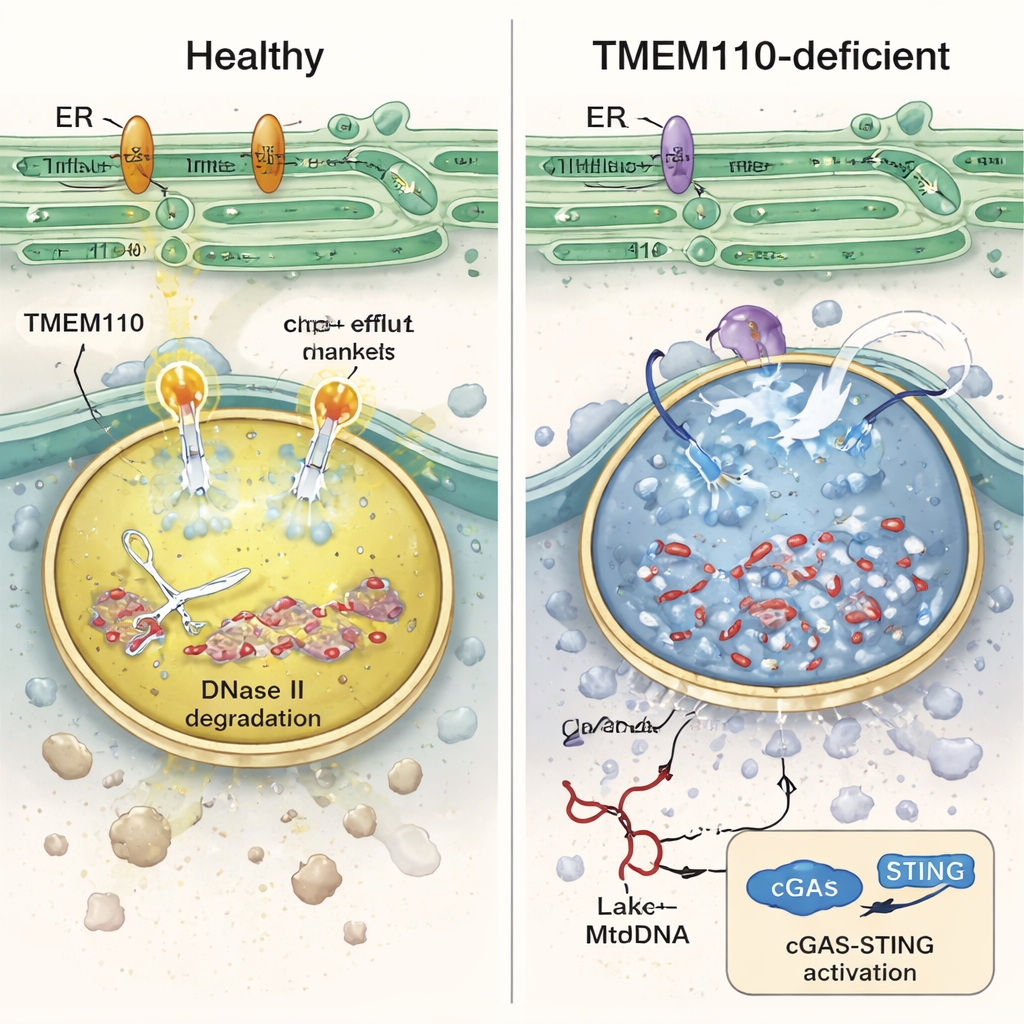
Sprzężenie zwrotne z systemem alarmowym DNA komórki
Gdy mtDNA przecieknie do cytosolu, rozpoznaje go ścieżka cGAS–STING, główny system alarmowy komórki dla zabłąkanego DNA. Co ciekawe, TMEM110 także wiąże się ze STING w spoczynku i utrzymuje go zaaresztowanego w siateczce śródplazmatycznej, zapobiegając niepotrzebnemu alarmowi. Gdy w cytosolu gromadzi się DNA, STING zostaje aktywowany i poluzowuje swoje więzy z TMEM110. To z kolei odsłania inną część TMEM110, która angażuje TRPML1 i zwiększa wypływ wapnia z lizosomów, tworząc pętlę sprzężenia zwrotnego: wykrycie własnego DNA aktywuje mechanizm, który wzmacnia jego usuwanie. Mutacje STING związane z chorobami, znalezione w ludzkich zespołach autoinflamacyjnych, zaburzają to przekazanie, uwięziając TMEM110 w stanie zahamowanym i przyczyniając się do przewlekłej aktywacji interferonu. U myszy niosących taką mutację STING, TBI prowadził do gorszych uszkodzeń narządów i wyższej śmiertelności, ale dostarczenie małego fragmentu TMEM110 do lizosomów za pomocą terapii mRNA–lipidowymi nanocząsteczkami częściowo przywróciło kontrolę nad mtDNA i poprawiło przeżywalność.
Wskazówki od pacjentów i ścieżki do nowych terapii
Następnie autorzy zwrócili się do kohorty klinicznej 143 osób z urazowymi uszkodzeniami mózgu i MODS. Pacjenci, których monocyty miały wyższe poziomy TMEM110 wczesno po urazie, mieli większe szanse na odzyskanie funkcji narządów i przeżycie. Ci z niższym TMEM110 wykazywali więcej krążącego mtDNA i zdecellularyzowanych mitochondriów, silniejsze odpowiedzi interferonowe, wyższe wyniki niewydolności narządów oraz szersze profile autoprzeciwciał, w tym przeciwciała przeciw DNA i białkom jądrowym. Wzorce te bardzo przypominały ustalenia u myszy, co wspiera tezę, że kontrola stabilności lizosomów i usuwania mtDNA napędzana przez TMEM110 jest wspólnym mechanizmem w chorobach ludzkich.
Co to oznacza dla pacjentów z ciężkimi urazami
Mówiąc prosto, praca ta sugeruje, że po ciężkim urazie nasz układ odpornościowy stoi przed delikatnym wyborem: cicho strawić zalew własnego DNA uwolnionego z umierających komórek, czy zinterpretować go jako inwazję wirusową i podjąć pełnoskalowy atak, który może uszkodzić własne narządy. TMEM110, poprzez regulację lizosomalnego kanału jonowego i współpracę z czujnikiem DNA STING, pomaga komórkom wybrać bezpieczniejszą ścieżkę. Gdy ten system zawodzi — z powodu braku TMEM110, mutacji STING lub przeciążenia lizosomów — mtDNA ucieka, alarm nie ustaje, a następnie następuje uszkodzenie wielu narządów. Celowanie w oś TMEM110–TRPML1–STING, na przykład za pomocą terapii mRNA skierowanych do lizosomów, może dać nową możliwość zapobiegania albo leczenia niewydolności narządów i powikłań przypominających autoimmunizację po ciężkich urazach.
Cytowanie: Feng, Z., Pan, Y., Zhou, J. et al. Tmem110 regulates the conformation of TRPML1 to maintain endolysosomal homeostasis and prevent mitochondrial DNA leakage and pathological self-DNA processing. Nat Commun 17, 1678 (2026). https://doi.org/10.1038/s41467-026-68382-8
Słowa kluczowe: urazowe uszkodzenie mózgu, mitochondrialne DNA, lizosomy, interferon typu I, ścieżka cGAS‑STING