Clear Sky Science · pl
Wykorzystanie liniowych odniesień z pangenomu do odkrywania brakujących wariantów autyzmu
Dlaczego ukryte zmiany DNA mają znaczenie w autyzmie
Większość rodzin, które decydują się na badania genetyczne u dziecka z autyzmem, liczy na jasne odpowiedzi, ale około cztery na pięć otrzymuje brak jednoznacznego wyjaśnienia genetycznego. To badanie zajmuje się kluczowym powodem tego stanu rzeczy: wiele istotnych zmian w DNA jest zbyt skomplikowanych, by wykryć je standardowymi testami. Tworząc niemal kompletne genomy dla 189 osób z 51 rodzin dotkniętych autyzmem i porównując je z nowym, bogatszym odniesieniem „pangenomu”, badacze pokazują, jak zaawansowane sekwencjonowanie może odsłonić rzadkie, wcześniej niewidoczne mutacje, które mogą pomóc wyjaśnić niektóre przypadki autyzmu i pokrewnych zaburzeń.
Patrząc poza standardowe testy genetyczne
Tradycyjne testy kliniczne opierają się na krótkich fragmentach DNA do skanowania genomu osoby. Dobrze sprawdzają się przy wielu jedno-literowych zmianach, ale często zawodzą w regionach powtarzalnych lub strukturalnie złożonych — dokładnie tam, gdzie kryją się niektóre silnie działające mutacje chorobotwórcze. Zespół skupił się na rodzinach, w których wcześniejsze testy oparte na krótkich odczytach genomu, egzomu lub panelach genowych nie wykryły przyczyny autyzmu lub objawów podobnych do zespołu Retta. Korzystając z sekwencjonowania długodystansowego, które odczytuje znacznie dłuższe odcinki DNA, zbudowali wysokiej jakości, fazowane złożenia genomów dla 189 osób. Oznacza to, że mogli odtworzyć dwie kopie każdego chromosomu u każdej osoby, jedną odziedziczoną od każdego rodzica, z bardzo niewielkimi brakami.
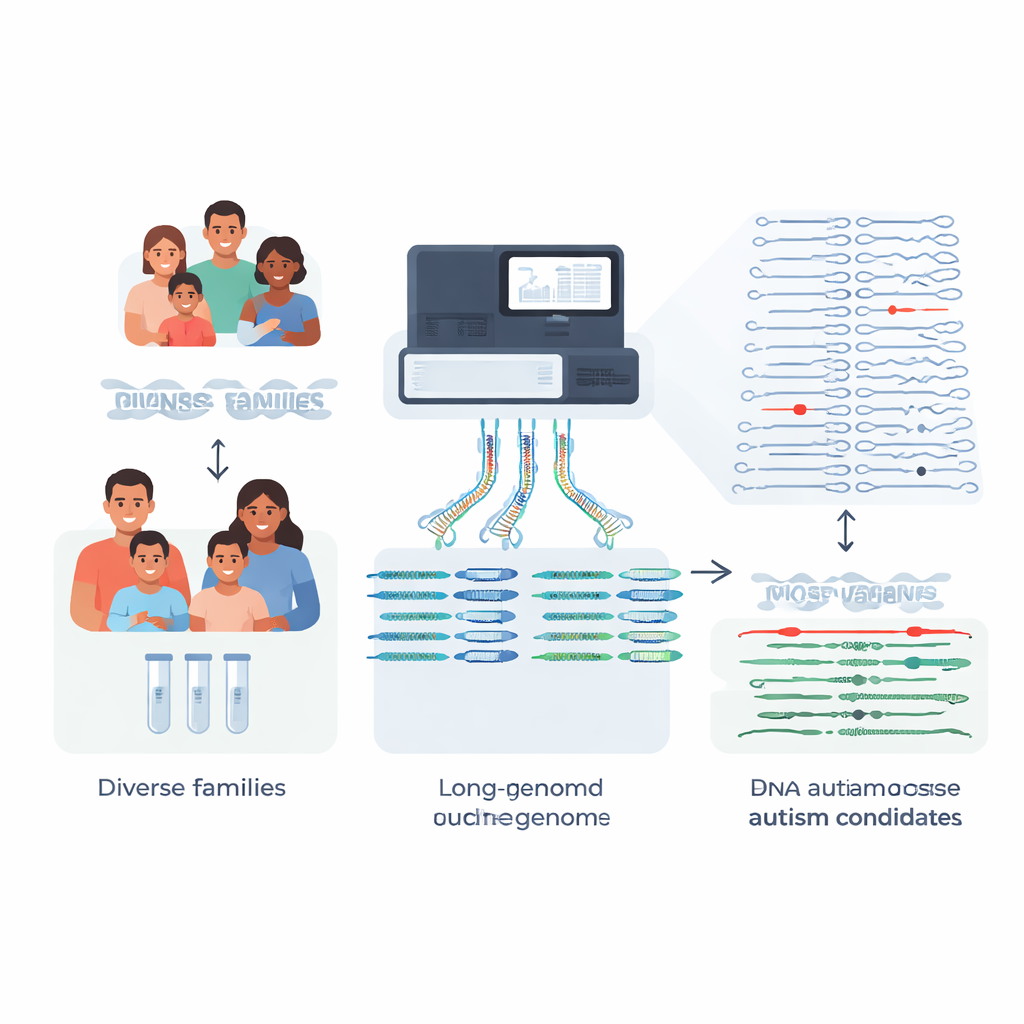
Warianty strukturalne: duże zmiany o dużych skutkach
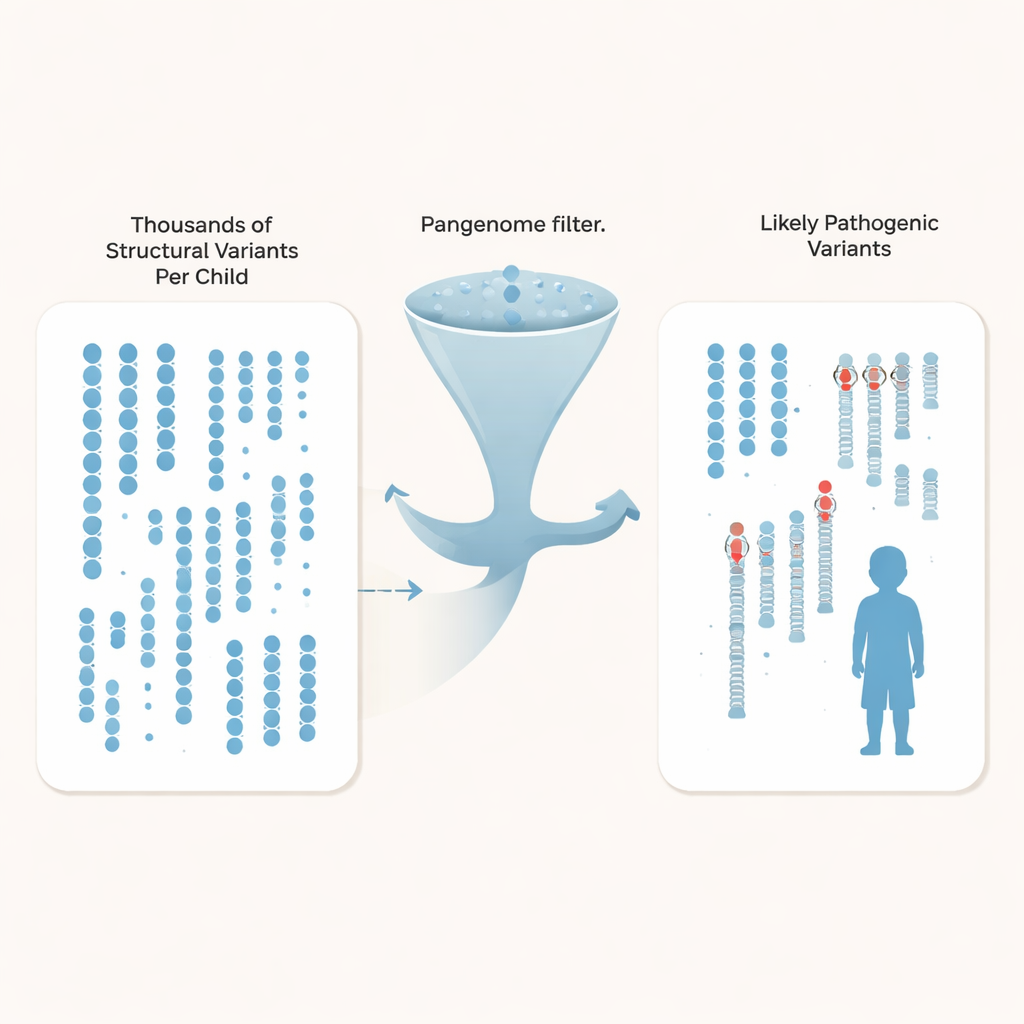
Zamiast ograniczać się do jedno-literowych różnic, badacze skupili się na wariantach strukturalnych — insercjach, delecjach i przestawieniach obejmujących co najmniej 50 nukleotydów, które mogą zaburzać geny lub ich elementy regulacyjne. Każde dziecko miało około 27 000 takich wariantów, ale zdecydowana większość to nieszkodliwe różnice tła występujące w populacji. Porównując rodziny z autyzmem z setkami głęboko zsekwencjonowanych genomów kontrolnych pangenomu pochodzących z różnych populacji, zespół mógł odfiltrować ponad 97% powszechnych wariantów strukturalnych u każdego dziecka, pozostawiając około 600 rzadkich kandydatów na genom, a w przypadku największego zestawu kontrolnego — nawet tylko około 200.
Odnalezienie pominiętych mutacji w znanych genach ryzyka
Gdy przestrzeń poszukiwań została drastycznie zawężona, autorzy połączyli kilka linii dowodów: znane geny związane z autyzmem i zaburzeniami neurorozwojowymi, regiony regulacyjne aktywne w rozwijającej się korze mózgowej oraz wzorce dziedziczenia w każdej rodzinie. Odkryli trzy jednoznacznie patogenne mutacje, których wcześniejsze testy nie wykryły. Był wśród nich nowy sygnał stop w genie SYNGAP1, ważnym dla funkcji synaps, oraz delecja usuwająca ostatni egzON genu MECP2, kluczowego w zespole Retta, mimo że pacjent przeszedł wiele wcześniejszych testów klinicznych. Potwierdzili też chorobotwórczą zmianę w TBL1XR1, genie współdziałającym z MECP2. Łącznie wyróżnili dziewięć dodatkowych wariantów strukturalnych — często dziedziczonych i zlokalizowanych w regionach regulacyjnych w pobliżu genów związanych z mózgiem — jako silne kandydatury do przyszłych badań funkcjonalnych.
Czego badanie nie wykazało — i dlaczego to nadal ma znaczenie
Mimo tak dogłębnych poszukiwań autorzy nie zaobserwowali wyraźnej ogólnej nadmiarowości wariantów strukturalnych u dzieci z autyzmem w porównaniu z ich nie dotkniętymi rodzeństwem, przynajmniej przy tak umiarkowanej liczebności próbki. Pojawiła się jednak sugestia większej liczby zmian strukturalnych na chromosomie X u dziewcząt z objawami, a niemal kompletne złożenia chromosomów X i Y pozwoliły zauważyć nietypowe wzorce, takie jak silne odchylenie inaktywacji chromosomu X. Te cechy mogą stać się ważnymi wskazówkami w miarę badania kolejnych rodzin. Kluczowe jest to, że praca pokazuje, iż sekwencjonowanie długodystansowe potrafi odzyskać patogenne warianty, które metody o krótkich odczytach pomijają — szczególnie w trudnych częściach genomu i w regionach kontrolnych precyzujących aktywność genów.
Co to oznacza dla rodzin i przyszłych badań
Dla rodzin bezpośredni wpływ jest umiarkowany, lecz istotny: wśród tych trudnych do rozwiązania przypadków około 6% uzyskało jasną diagnozę genetyczną, a prawie jedna na pięć rodzin otrzymała silne nowe kandydatury wariantów do zbadania. Dla całej dziedziny przesłanie jest większe. W miarę dodawania do pangenomu bardziej zróżnicowanych, kompletnych genomów i upowszechniania sekwencjonowania długodystansowego, klinicyści będą mogli wykluczyć powszechne zmiany strukturalne i szybko skupić się na niewielkim zestawie rzadkich, potencjalnie szkodliwych wariantów u każdego pacjenta. Ta zmiana może stopniowo przekształcić dzisiejsze liczne „nierozwiązane” przypadki autyzmu w takie, gdzie podstawowa biologia — i możliwe drogi wsparcia oraz leczenia — będą znacznie lepiej rozumiane.
Cytowanie: Sui, Y., Lin, J., Noyes, M.D. et al. Using the linear references from the pangenome to discover missing autism variants. Nat Commun 17, 1681 (2026). https://doi.org/10.1038/s41467-026-68378-4
Słowa kluczowe: genetyka autyzmu, sekwencjonowanie długodystansowe, warianty strukturalne, ludzki pangenom, zespół Retta