Clear Sky Science · pl
Molekularne podstawy antagonizmu dimerowego ludzkiego receptora wazopresyny 1A
Dlaczego receptor hormonalny w mózgu ma znaczenie
Hormony takie jak wazopresyna i oksytocyna są najbardziej znane z regulacji bilansu wodnego, ciśnienia krwi, porodu i więzi społecznych. Jednak sposób działania ich receptorów na poziomie atomowym pozostawał w dużej mierze ukryty. Niniejszy artykuł ujawnia szczegółowe struktury 3D jednego kluczowego receptora — ludzkiego receptora wazopresyny V1a — który wiązany jest z zachowaniami społecznymi, reakcją na stres i kilkoma zaburzeniami mózgu. Zrozumienie jego kształtu i mechanizmu blokowania przez leki może pomóc naukowcom w projektowaniu lepszych terapii dla schorzeń takich jak autyzm, zespół stresu pourazowego (PTSD) czy choroba Huntingtona.
Bliźniaczy receptor kształtujący sygnały serca, nerek i mózgu
Receptor V1a znajduje się na powierzchni wielu komórek w organizmie, szczególnie w naczyniach krwionośnych, nerkach i niektórych obszarach mózgu. Gdy hormon wazopresyna wiąże się z receptorem, uruchamiane są wewnętrzne szlaki sygnałowe kontrolujące ciśnienie krwi, równowagę płynów oraz obwody mózgowe związane z interakcjami społecznymi, emocjami i stresem. Badania genetyczne i kliniczne powiązały nieprawidłowe sygnalizowanie V1a z zaburzeniami ze spektrum autyzmu, PTSD oraz chorobą Huntingtona, co czyni go atrakcyjnym celem terapeutycznym. Istnieje kilka leków blokujących V1a (antagonistów), które są już stosowane lub testowane klinicznie, jednak dotąd nikt nie widział ludzkiego receptora V1a w wysokiej rozdzielczości, pozostawiając poważne pytania dotyczące sposobu jego składania i dokładnego mechanizmu unieszkodliwiania przez te związki. 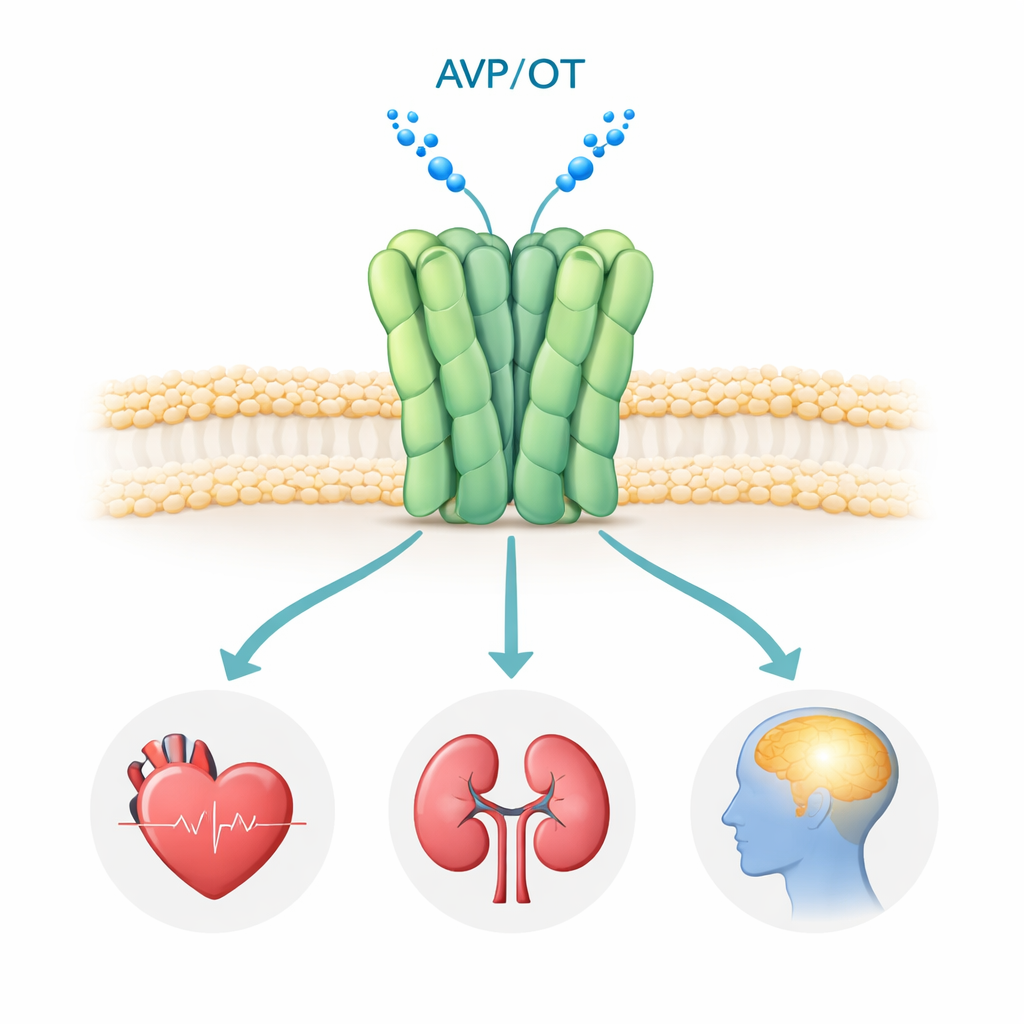
Uchwycenie struktury receptora w wielu stanach związanych z lekami
Naukowcy zastosowali krio-elektronową mikroskopię (cryo–EM), technikę, która zamraża białka błyskawicznie i obrazuję je wiązką elektronów, aby zobrazować ludzki receptor V1a. Aby ustabilizować białko, zaprojektowali nieco zmodyfikowaną formę, która nadal dobrze wiąże leki, i połączyli ją z małym fragmentem przeciwciała (nanociałkiem) ułatwiającym obrazowanie. Rozwiązali struktury receptora zarówno w stanie wolnym, jak i związanym z trzema medycznie istotnymi antagonistami: atosibanem (peptydowym lekiem stosowanym w zapobieganiu przedwczesnym porodom) oraz dwoma małocząsteczkowymi związkami przenikającymi do mózgu — balovaptanem i SRX246 — które były testowane u osób z autyzmem lub chorobą Huntingtona. Wszystkie struktury osiągnęły niemal atomową rozdzielczość, ujawniając położenie siedmiu helis przechodzących przez błonę receptora, elastycznych pętli i związanych z nimi leków.
Receptor, który woli pracować w parach
W przeciwieństwie do spokrewnionych receptorów, widzianych dotąd jedynie jako jednostki monomeryczne, V1a pojawił się jako para — dimer — we wszystkich czterech strukturach cryo-EM. Dwa receptory leżą obok siebie w błonie, nawiązując ciasne kontakty głównie przez jedną z ich helis, wspomagane zarówno przez kontakty polarne, jak i hydrofobowe. Aby sprawdzić, czy takie parowanie zachodzi również w żywych komórkach, zespół złączył V1a z jasnym białkiem fluorescencyjnym i użył metody fotoblokowania pojedynczych cząsteczek: jeśli punkt na powierzchni komórki zawierał dwie kopie receptora, jego sygnał światła zanikałby w dwóch krokach. Około trzy czwarte obserwowanych punktów bledło dokładnie w dwóch etapach, co silnie popiera ideę, że V1a naturalnie tworzy dimery na powierzchni komórkowej. Gdy naukowcy zmienili kluczowe reszty kontaktowe, aby zaburzyć interfejs, receptor przesunął się w stronę jednostek pojedynczych i stał się mniej wrażliwy na hormon i lek, co sugeruje, że dimer nie jest tylko elementem strukturalnym, lecz ma znaczenie funkcjonalne. 
Elastyczna bramka przy wejściu dla hormonu
Zespół odkrył nieoczekiwany region „bramki” nazwany zewnętrzną pętlą 2 (ECL2), która leży u wierzchołka kieszeni wiązania hormonu. W stanie bez leku (apo) ta pętla rozciąga się płasko nad kieszenią jak pokrywa i nie tworzy zwykłego mostka dwusiarczkowego (wiązania siarka–siarka) obserwowanego w wielu spokrewnionych receptorach. Zamiast tego fragmenty pętli składają się do wnętrza kieszeni i są utrzymywane przez sieć oddziaływań z otaczającymi helisami, częściowo zasłaniając dużą, lepko-przyciągającą jamę wiążącą. Gdy którykolwiek z trzech antagonistów się wiąże, ECL2 unosi się i odsuwa, tworząc klasyczny mostek dwusiarczkowy oraz szeroką, wypełnioną rozpuszczalnikiem jamę, którą zajmują leki. Ten dramatyczny ruch sugeruje, że V1a może używać ECL2 jako dynamicznej bariery ograniczającej przypadkową aktywację przez obce cząsteczki, a leki można projektować albo tak, by uwięzić pętlę w płaskim „stanie spoczynkowym”, albo by wykorzystać jej podniesioną, otwartą konformację.
Jak trzy leki wyciszają ten sam receptor na różne sposoby
Atosiban, który bardzo przypomina naturalny hormon oksytocynę, rozciąga się od wierzchołka kieszeni aż do jej dna, kotwicząc się za pomocą kombinacji wiązań wodorowych i kontaktów hydrofobowych. Poprzez zmianę kilku kluczowych pozycji w porównaniu z oksytocyną, nie wywołuje łańcucha wewnętrznych przesunięć zwykle wymaganych do aktywacji receptora: kluczowe reszty „mikroprzełączników”, które poruszają się w trakcie sygnalizacji, pozostają zablokowane w swoich nieaktywnych pozycjach, wewnętrzna jama umożliwiająca przyjęcie białka G nigdy się nie otwiera, a ważne dla aktywacji miejsce wiążące magnez jest zaburzone. W przeciwieństwie do tego balovaptan i SRX246 to kompaktowe, niepeptydowe cząsteczki, które wnikają głęboko w kieszeń, ale stosują odmienne strategie. Balovaptan opiera się na sztywnym, hydrofobowym rdzeniu, który ciasno wypełnia głęboki wcięcie, oraz elastycznym, polarnym ogonie sięgającym w kierunku wejścia kieszeni. SRX246 wykorzystuje modułową, fragmentopodobną architekturę kotwiczoną przez rdzeń β-laktamowy, z różnymi „strefami” wypełniającymi subkieszonki i sięgającymi w stronę pętli zewnątrzkomórkowych. W obu przypadkach leki stabilizują nieaktywną konformację niekompatybilną z wiązaniem białka G. Subtelne różnice w kształcie i chemii kieszeni — zwłaszcza w dwóch pozycjach na helisach 5 i 7 — pomagają wyjaśnić, dlaczego balovaptan i SRX246 wykazują preferencję wobec V1a nad blisko spokrewnionymi receptorami.
Implikacje dla przyszłych terapii
Dostarczając zdjęć o wysokiej rozdzielczości V1a jako dimeru, ujawniając wcześniej niewidzianą „płaską” konformację pętli w stanie bez leku oraz szczegółowo opisując, jak trzy bardzo różne antagonisty wyłączają receptor, ta praca daje projektantom leków precyzyjną mapę strukturalną do ukierunkowania V1a. Sugeruje sposoby opracowania leków następnej generacji, które albo wykorzystają cechy specyficzne dla dimeru, albo zablokują receptor w szczególnie nieaktywnym stanie podstawowym, z ostatecznym celem leczenia zaburzeń mózgu i związanych ze stresem bardziej selektywnie i z mniejszą liczbą skutków ubocznych.
Cytowanie: Zhong, P., Chu, B., Yu, Z. et al. Molecular basis of antagonism of the dimeric human arginine vasopressin receptor 1A. Nat Commun 17, 1622 (2026). https://doi.org/10.1038/s41467-026-68331-5
Słowa kluczowe: receptor wazopresyny V1a, receptor sprzężony z białkiem G, dimeryzacja receptora, struktura cryo-EM, projektowanie leków neuropsychiatrycznych