Clear Sky Science · pl
Glicerol 3‑fosforan acylotransferaza nasila toksyczność indukowaną przez α‑synukleinę przez zwiększenie peroksydacji lipidów
Dlaczego tłuszcze w mózgu mają znaczenie dla Parkinsona
Chorobę Parkinsona zwykle opisuje się jako problem z białkiem zwanym alfa‑synukleiną, które zlepia się i uszkadza komórki mózgowe kontrolujące ruch. To badanie pokazuje, że tłuszcze mózgowe — szczególnie sposób ich powstawania i uszkadzania — odgrywają zaskakująco istotną rolę w tym, jak toksyczna staje się alfa‑synukleina. Identyfikując enzym odpowiadający za tworzenie lipidów, który pogarsza uszkodzenie neuronów, praca wskazuje nową, podatną na leki ścieżkę, mogącą uzupełnić dotychczasowe wysiłki ukierunkowane na źródła Parkinsona.
Białko, które źle się zachowuje w Parkinsonie
Osoby z chorobą Parkinsona stopniowo tracą neurony produkujące dopaminę w głębokiej części mózgu odpowiedzialnej za koordynację ruchu. W umierających komórkach naukowcy często znajdują gęste złogi zwane ciałami Lewy’ego, wypełnione białkiem alfa‑synukleiną. W rzadkich rodzinach mutacje lub dodatkowe kopie genu alfa‑synukleiny bezpośrednio wywołują Parkinsona, lecz powszechne warianty tego genu zwiększają ryzyko jedynie umiarkowanie. To sugeruje, że inne geny i szlaki modyfikują, jak szkodliwa jest alfa‑synukleina. Coraz więcej dowodów wskazuje na lipidy — tłuszcze i cząsteczki podobne do tłuszczów, które tworzą błony komórkowe i magazyny energii — jako kluczowych partnerów zarówno w zlepianiu alfa‑synukleiny, jak i w śmierci neuronów.
Odkrycie silnego enzymu lipidowego w modelach muszek owocówek
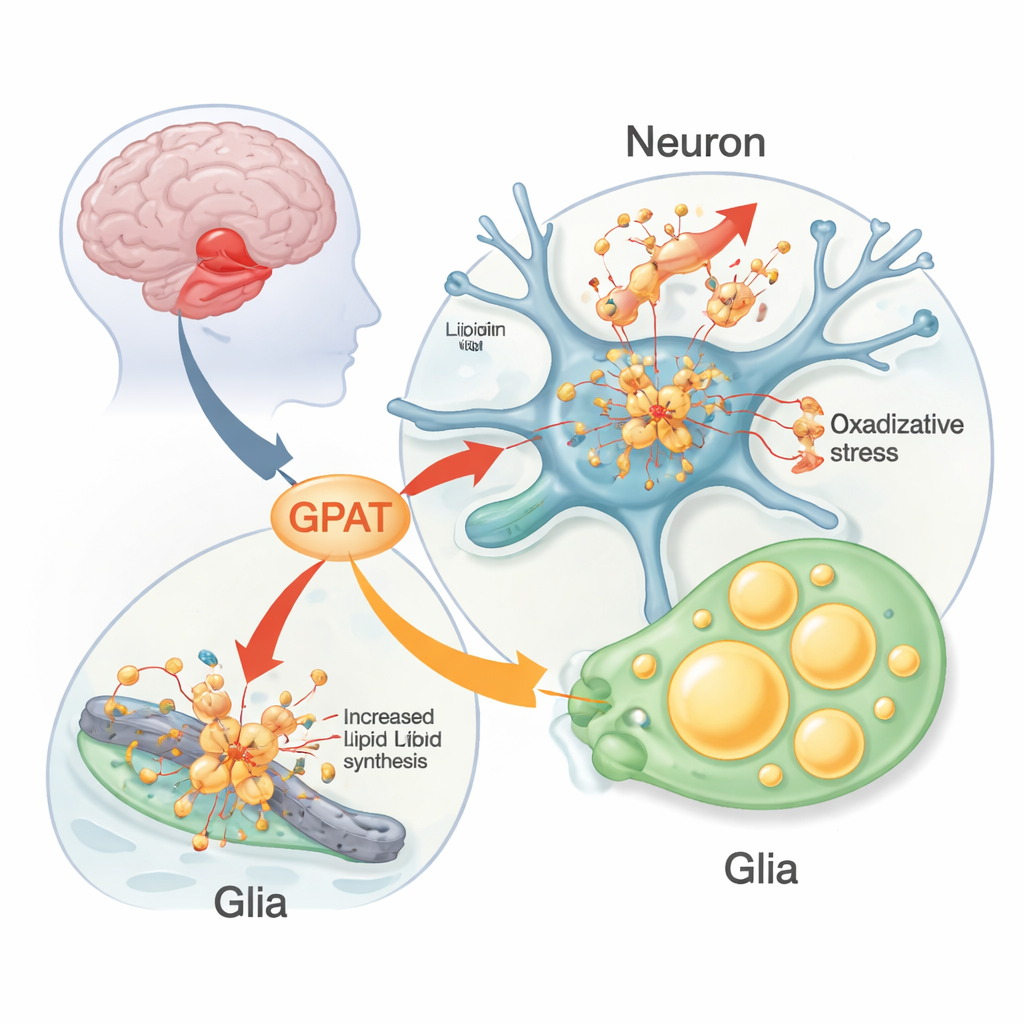
Naukowcy wykorzystali muszki owocówki zmodyfikowane tak, by w ich układzie nerwowym wytwarzano ludzką alfa‑synukleinę jako żywe laboratorium badań. Muszki te rozwijają objawy podobne do Parkinsona: utratę neuronów produkujących dopaminę, problemy z wchodzeniem po ściance i zaburzenia rytmów aktywności dobowej. Zespół systematycznie modyfikował musze wersje ludzkich genów ryzyka Parkinsona, by sprawdzić, które z nich zmieniają wpływ alfa‑synukleiny. Wyróżnił się jeden gen: mino, kodujący mitochondrialną formę enzymu glicerol 3‑fosforan acylotransferazy (GPAT). GPAT działa na wejściu do syntezy fosfolipidów i trójglicerydów — lipidów tworzących błony i krople tłuszczu. Kiedy zespół zmniejszył aktywność mino w neuronach, muszki z alfa‑synukleiną zachowały więcej neuronów dopaminergicznych i dłużej poruszały się lepiej; zwiększenie aktywności mino miało przeciwny, szkodliwy efekt.
Uszkodzone tłuszcze, zestresowane mitochondria i krople tłuszczu w gleju
Zgłębiając temat, naukowcy odkryli, że GPAT wpływa na kumulację uszkodzeń oksydacyjnych w lipidach mózgu. U muszek z alfa‑synukleiną utrzymywanych w wyższej temperaturze (co nasila cechy choroby) peroksydacja lipidów — chemiczne „rdzewienie” tłuszczów — wzrastała w błonach mózgowych. Obniżenie aktywności mino ograniczało to uszkodzenie, natomiast jego nadeksresja je zwiększała; bez alfa‑synukleiny zmiany mino miały niewielki efekt. Markery śmierci komórek w obszarze wzrokowym mózgu odzwierciedlały ten sam wzór. Zespół zaobserwował także uderzające nagromadzenie kropli lipidowych — maleńkich sfer magazynujących tłuszcz — nie w neuronach, lecz w sąsiednich komórkach glejowych. Krople te powiększały się z wiekiem u muszek z alfa‑synukleiną i były modyfikowane przez enzymy syntetyzujące lub rozkładające trójglicerydy, co podkreśla aktywne metaboliczne partnerstwo między neuronami a glejem pod stresem.
Przebudowa metabolizmu i zlepianie się alfa‑synukleiny
Pomiary metabolitów z mózgów muszek wykazały, że ekspresja alfa‑synukleiny wiązała się z zatorami w cyklu wytwarzania energii komórki: cytrynian i izocytrynian, dwa pośrednie związki cyklu kwasu trójkarboksylowego (TCA), gromadziły się silnie, podczas gdy dalsze etapy przesuwały się mniej intensywnie. Poziomy mleczanu również wzrastały, co jest zgodne z nasiloną glikolizą. Jednocześnie szczegółowe profilowanie lipidów wykazało zmiany w równowadze fosfolipidów błonowych i ich składu kwasów tłuszczowych, z przewagą gatunków bardziej podatnych na uszkodzenia oksydacyjne. Gdy zespół zmniejszył aktywność kilku enzymów GPAT — mino w mitochondriach oraz powiązanych enzymów w retikulum endoplazmatycznym — alfa‑synukleina wciąż się gromadziła, lecz jej skłonność do tworzenia oligomerów wyższych rzędów (wielobiałkowych skupień) spadła, a mitochondria wykazywały mniej oznak stresu oksydacyjnego i „starzenia”.
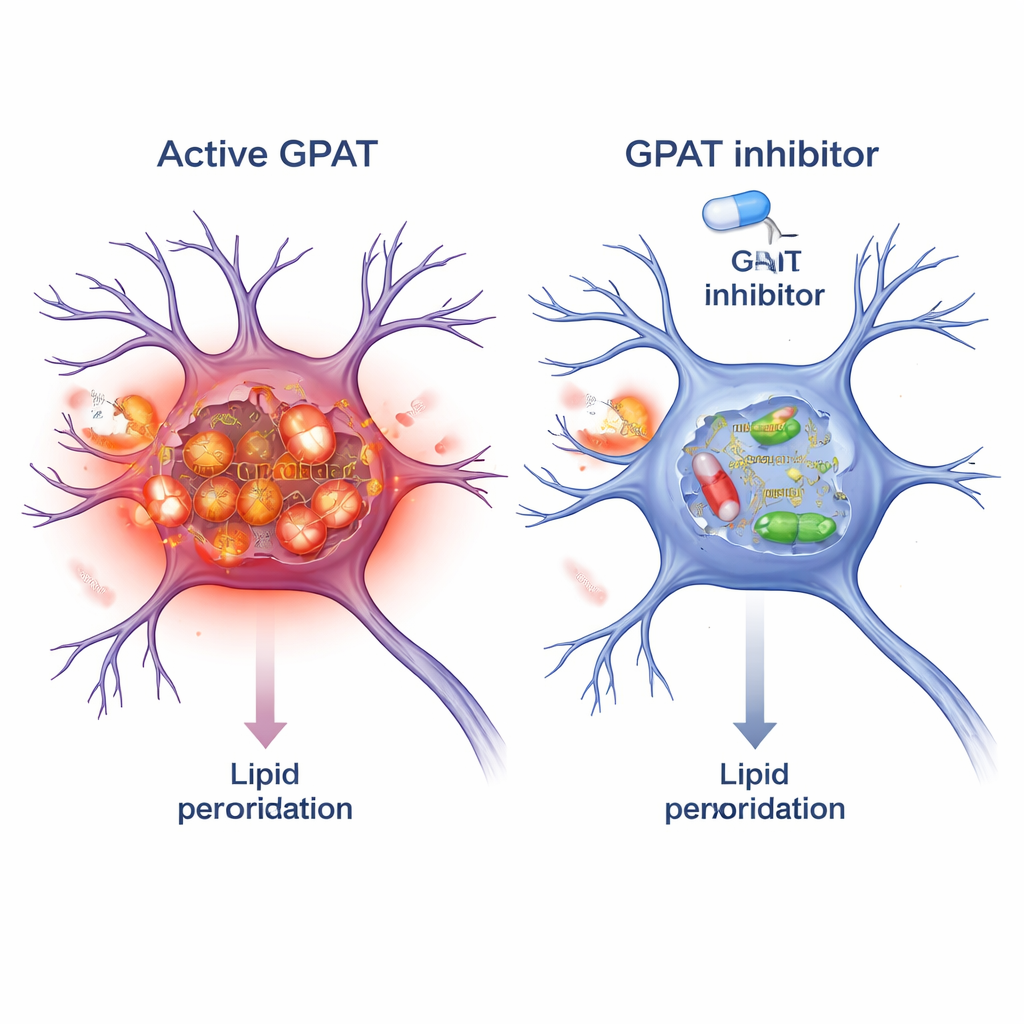
Blokowanie GPAT jako strategia ochronna
Ponieważ GPAT jest enzymem, można go zaatakować małocząsteczkowymi lekami. Badacze przetestowali FSG67, istniejący inhibitor GPAT początkowo opracowany w kontekście otyłości i cukrzycy. U muszek z alfa‑synukleiną podawanie FSG67 w pokarmie odtworzyło korzyści genetycznego wyciszenia GPAT: poprawę ruchu, lepsze przeżycie neuronów dopaminergicznych, mniej szkodliwych oligomerów alfa‑synukleiny i zmniejszony stres oksydacyjny mitochondriów. Aby sprawdzić, czy ten pomysł przełoży się na ssaki, potraktowali hodowane neurony myszy wcześniej utworzonymi fibrylami alfa‑synukleiny, które nasionują toksyczne agregaty. Wspoleczenie z FSG67 zmniejszyło gromadzenie fosforylowanej alfa‑synukleiny i obniżyło wiele niezależnych markerów peroksydacji lipidów w tych neuronach.
Co to oznacza dla osób z chorobą Parkinsona
Mówiąc wprost, praca ta pokazuje, że sposób, w jaki mózg gospodaruje tłuszczami, może przygasać albo nasilać toksyczność alfa‑synukleiny. Gdy GPAT jest silnie aktywny, do błon i kropli magazynowych wbudowywane są bardziej wrażliwe lipidy, łatwiejsze do utlenienia; to uszkodzone środowisko lipidowe sprzyja pojawianiu się szkodliwych form alfa‑synukleiny i obciąża mitochondria, elektrownie komórkowe. Ograniczenie aktywności GPAT — genetycznie lub za pomocą leku — przesuwa równowagę w stronę mniejszej „rdzy” lipidów, mniejszej liczby toksycznych agregatów białkowych i zdrowszych neuronów. Choć wyniki są wstępne i pochodzą z muszek oraz hodowanych komórek mysich, zwracają uwagę na metabolizm lipidów, a w szczególności na GPAT, jako obiecujący nowy kierunek terapii choroby Parkinsona, który mógłby uzupełniać strategie bezpośrednio ukierunkowane na alfa‑synukleinę.
Cytowanie: Ren, M., Lim, G.G.Y., Tang, W. et al. Glycerol 3-phosphate acyltransferase exacerbates α-synuclein-induced toxicity by increasing lipid peroxidation. Nat Commun 17, 1618 (2026). https://doi.org/10.1038/s41467-026-68325-3
Słowa kluczowe: Choroba Parkinsona, alfa‑synukleina, peroksydacja lipidów, inhibitor GPAT, neurodegeneracja