Clear Sky Science · pl
Redukcja RAD23A wydłuża życie i łagodzi patologię w mysich modelach proteinopatii TDP-43
Dlaczego te badania są ważne dla rodzin i pacjentów
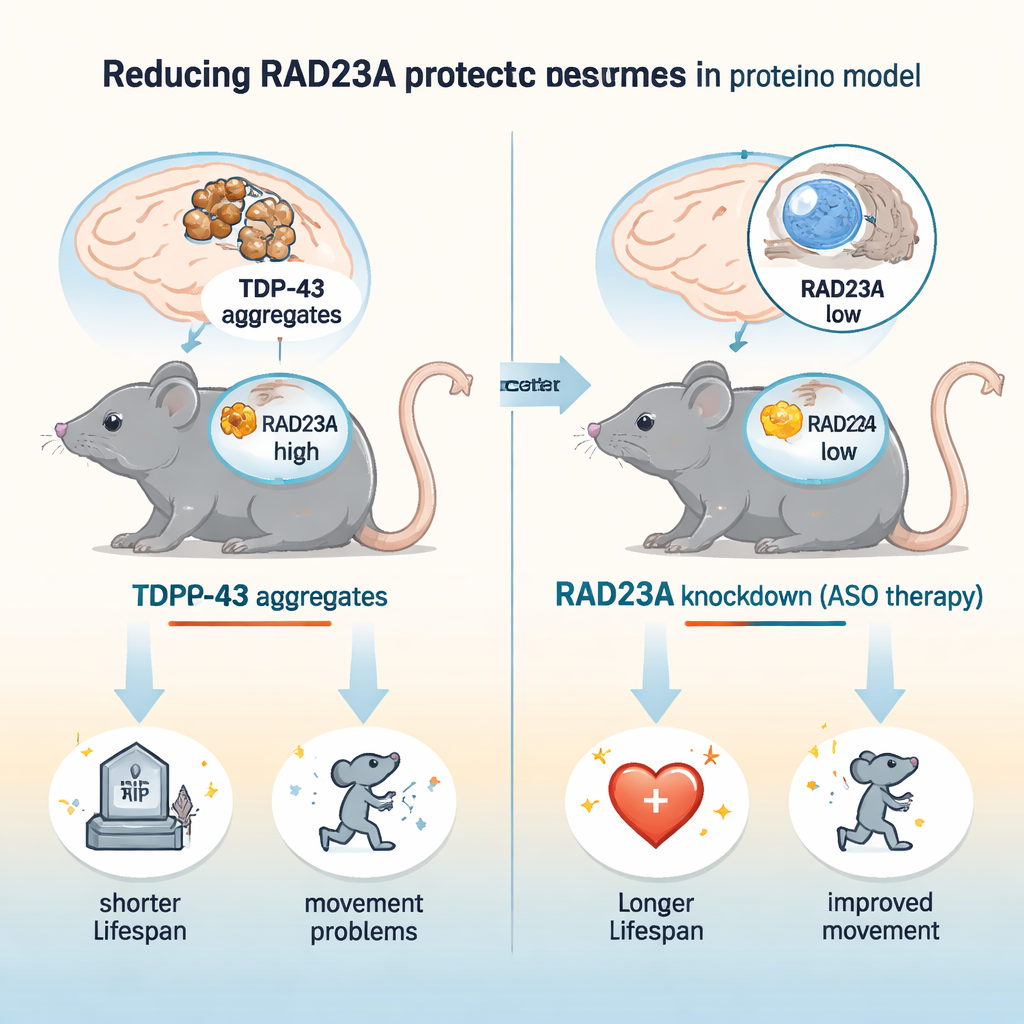
Wiele postaci demencji i chorób neuronów ruchowych, w tym stwardnienie zanikowe boczne (ALS) i otępienie czołowo-skroniowe (FTD), wiąże się z białkami w komórkach mózgu, które nieprawidłowo się fałdują, agregują i stopniowo zatruwają neurony. Jednym z kluczowych sprawców jest białko TDP-43, które normalnie pomaga w regulacji RNA, ale staje się toksyczne, gdy tworzy agregaty. W tym badaniu postawiono nadzieję, że można uczynić komórki mózgowe bardziej odpornymi, ograniczając działanie innego białka, RAD23A, które bierze udział w obsłudze uszkodzonych białek. Autorzy pokazują na myszach, że obniżenie poziomu RAD23A może wydłużyć życie, poprawić ruch oraz zmniejszyć uszkodzenia mózgu w modelu choroby napędzanej przez TDP-43, co sugeruje nową strategię terapeutyczną.
Korek w transporcie białek w chorych neuronach
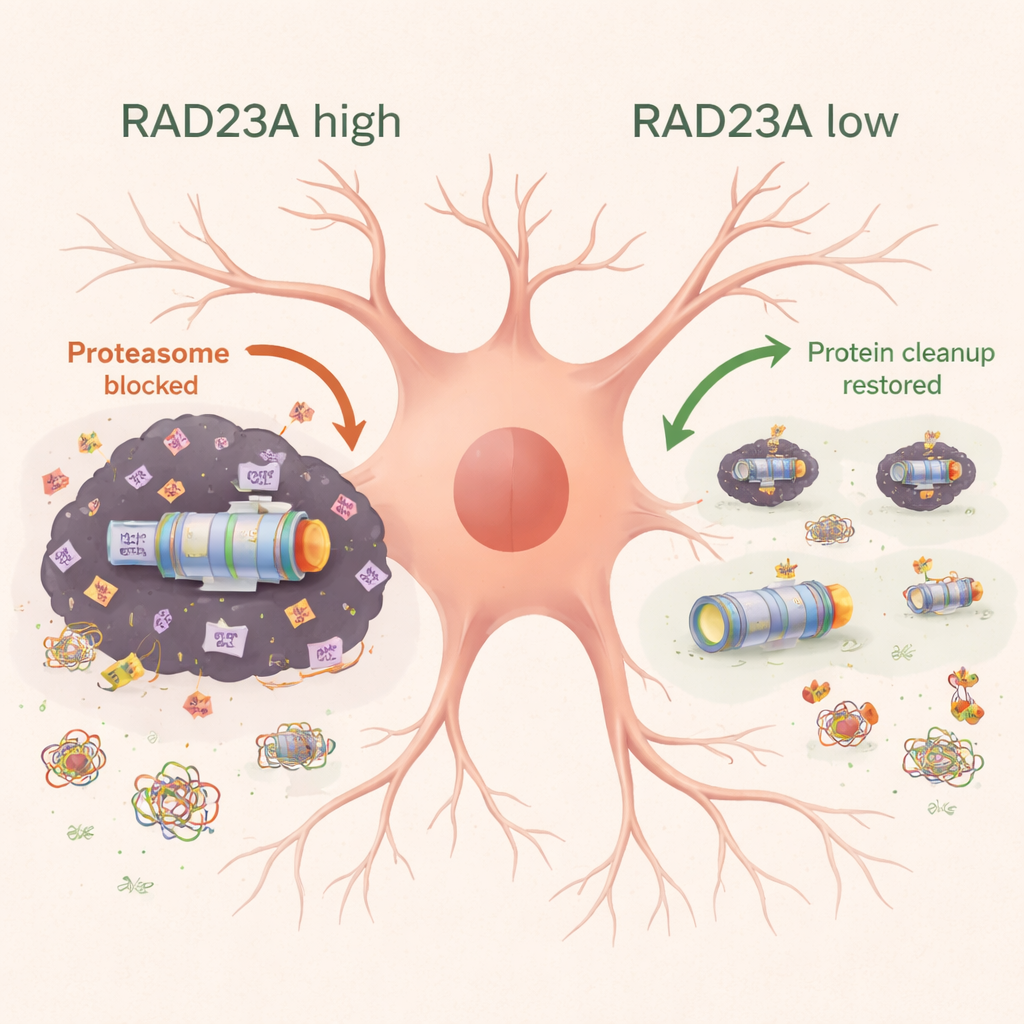
Choroby neurodegeneracyjne często charakteryzują się nagromadzeniem nieprawidłowo sfałdowanych białek, których komórkowe mechanizmy utylizacji nie są w stanie usunąć. W ALS i FTD TDP-43 opuszcza jądro komórkowe, tworzy lepkie grudki i silnie ulega ubikwitynacji — sygnałowi, który normalnie kieruje białka do proteasomu, głównego „niszczarki” komórki. RAD23A jest jednym z kilku białek‑„przewoźników”, które mogą dostarczać ubikwitynowane ładunki do proteasomu. Wcześniejsze badania na nicieniach i hodowlach neuronów sugerowały jednak paradoksalnie, że utrata białek podobnych do RAD23 może chronić przed uszkodzeniem wywołanym przez TDP-43 — paradoks, który to badanie postanowiło zbadać w żywym mózgu ssaka.
Przyciszanie RAD23A w modelu myszy z TDP-43
Naukowcy użyli dobrze opisanej mysiej linii, zwanej TAR4/4, która nadmiernie produkuje ludzkie TDP-43 w neuronach i rozwija zaburzenia ruchu, skoliozę, drżenie oraz przedwczesną śmierć, odzwierciedlając kluczowe cechy ALS/FTD. Obniżali RAD23A na dwa sposoby: poprzez wstrzyknięcie nowo narodzonym myszom antysensownych oligonukleotydów (ASO), które zmniejszają poziom RNA Rad23a, oraz przez krzyżowanie z myszami noszącymi genetyczne wyłączenie Rad23a. Jednorazowe leczenie ASO zmniejszyło poziomy RAD23A w mózgu i rdzeniu kręgowym o około trzy czwarte. U tych myszy z TDP-43 redukcja RAD23A wydłużyła żywotność o około 50% i opóźniła początek oraz zmniejszyła nasilenie zaburzeń chodu, drżeń, skrzywienia kręgosłupa i zaciśnięcia kończyn tylnych. Co ciekawe, całkowita genetyczna utrata RAD23A nie przyniosła dodatkowej korzyści, co sugeruje, że częściowe obniżenie jest optymalne, a długotrwały, całkowity brak może wywołać zmiany kompensacyjne.
Mniej zapalenia, lepsza obsługa białek i spokojniejsze genom
Badanie mikroskopowe kory ruchowej wykazało, że myszy z TDP-43 traciły neurony i wykazywały silną aktywację astrocytów i mikrogleju — komórek wspierających i odpornościowych mózgu. Obniżenie RAD23A zachowało liczbę neuronów i zmniejszyło markery zapalenia oraz śmierci komórek. Analizy biochemiczne ujawniły, że nadprodukcja TDP-43 zalała komórki ubikwitynowanymi, detergent‑nierozpuszczalnymi białkami i wciągnęła podjednostki proteasomu do tych agregatów, osłabiając zdolność komórki do usuwania uszkodzonych białek. Redukcja RAD23A zmniejszyła całkowite obciążenie ubikwitynowanymi białkami, utrzymała więcej proteasomów w rozpuszczalnej, czynnej puli i przywróciła kilka rodzajów aktywności proteasomu w kierunku normy. Równocześnie uderzenie w RAD23A zmniejszyło zarówno całkowite, jak i zaggregowane formy TDP-43, w tym szczególnie toksyczny fragment o masie ~25 kilodaltonów, i przesunęło TDP-43 z cytoplazmy z powrotem w kierunku jądra. Sekwencjonowanie RNA całego genomu pokazało, że tysiące zmian ekspresji genów wywołanych przez TDP-43 zostało częściowo odwróconych po redukcji RAD23A, zwłaszcza geny zaangażowane w funkcjonowanie neuronów, produkcję energii w mitochondriach i szlaki usuwania agregatów, takie jak agrefagia.
Przebudowa ukrytego „nierozpuszczalnego” proteomu
Aby przyjrzeć się bliżej uporczywym agregatom opornym na zwykłe detergenty, zespół użył spektrometrii mas z ciężkimi izotopami, aby skatalogować białka uwięzione w frakcji nierozpuszczalnej kory mózgowej myszy. Ekspresja ludzkiego TDP-43 przyciągała składniki proteasomu, białka cytoszkieletu i transportu oraz inne elementy maszynerii komórkowej. Gdy RAD23A zostało stłumione, ogólny skład tego nierozpuszczalnego proteomu uległ zmianie: mniej białek proteasomu i związanych z transportem było sekwestrowanych, podczas gdy niektóre białka rybosomalne i związane ze stresem wzrosły w agregatach. Co ważne, ta przebudowa nie odzwierciedlała po prostu zmian w poziomach RNA, co sugeruje, że RAD23A wpływa głównie na to, jak istniejące białka są dzielone między stany rozpuszczalne i zaggregowane, a nie na to, ile danego białka jest syntetyzowane.
Co to może znaczyć dla przyszłych terapii
Łącznie te wyniki przedstawiają RAD23A jako silny regulator kontroli jakości białek w neuronach pod stresem. Częściowe obniżenie RAD23A w mysim modelu napędzanym przez TDP-43 pozwoliło zmniejszyć toksyczne grudki białkowe, przywrócić aktywność maszynerii do usuwania białek, złagodzić szkodliwe zmiany w ekspresji genów, ograniczyć zapalenie mózgu oraz wydłużyć życie i funkcje motoryczne. Ponieważ nieprawidłowe nagromadzenie TDP-43 jest powszechne zarówno w postaciach dziedzicznych, jak i sporadycznych ALS, FTD i pokrewnych zaburzeń, ukierunkowanie RAD23A przy użyciu antysensownych leków kompatybilnych z ludźmi może oferować sposób ochrony neuronów bez bezpośredniego blokowania samego TDP-43, który pełni ważne funkcje. Choć wciąż wiele trzeba przetestować w innych modelach i u ludzi, praca ta wskazuje RAD23A jako obiecującą dźwignię w powszechnym szlaku neurodegeneracji.
Cytowanie: Guo, X., Prajapati, R.S., Chun, J. et al. Reduction of RAD23A extends lifespan and mitigates pathology in a mouse model of TDP-43 proteinopathy. Nat Commun 17, 1820 (2026). https://doi.org/10.1038/s41467-025-65104-4
Słowa kluczowe: TDP-43, ALS, agregacja białek, proteasom, terapia antysensowna