Clear Sky Science · pl
Najlepsze praktyki i narzędzia w R i Pythonie do przetwarzania statystycznego i wizualizacji danych lipidomiki i metabolomiki
Dlaczego ważne jest zamienianie laboratoryjnych liczb w czytelne obrazy
Nowoczesne instrumenty potrafią dziś zmierzyć tysiące drobnych cząsteczek — lipidów i innych metabolitów — w jednej kropli krwi lub tkanki. Te pomiary zawierają wskazówki dotyczące ryzyka chorób, odpowiedzi na leczenie oraz tego, jak nasze ciało reaguje na dietę czy starzenie. Jednak surowy wynik to nie gotowa odpowiedź: to ogromna tabela liczb, którą trzeba oczyścić, przeanalizować i przekształcić w zrozumiałe wizualizacje. Artykuł wyjaśnia, jak badacze mogą używać dwóch popularnych języków programowania, R i Python, by robić to rzetelnie, przejrzyście i z grafiką nadającą się do publikacji.
Od pomiarów chemicznych do złożonych tabel danych
W lipidomice i metabolomice spektrometria mas oraz chromatografia generują duże zbiory danych, w których każdy wiersz to próbka, a każda kolumna to cząsteczka. Te tabele rzadko zachowują się jak porządne przykłady z podręcznika. Zawierają wartości brakujące, obserwacje odstające i rozkłady skośne, gdzie kilka związków wykazuje ekstremalnie wysokie stężenia. Stężenia mogą rozciągać się na wiele rzędów wielkości i być wpływane przez wiek, płeć, dietę, leki, rytmy dobowy oraz czynniki techniczne, takie jak dryf aparatu czy efekty partii. Międzynarodowe grupy ekspertów wydały wytyczne standaryzujące sposób pobierania, przetwarzania i raportowania próbek, ale nawet przy dobrej praktyce laboratoryjnej niezbędne jest staranne przetwarzanie statystyczne, by wydobyć prawdziwe sygnały biologiczne z tego zaszumionego tła.
Czyszczenie i przygotowanie liczb
Zanim jakiekolwiek porównanie między grupami zdrowymi i chorymi nabierze sensu, dane trzeba przygotować. Przegląd opisuje, jak powstają wartości brakujące — w wyniku losowych awarii, ograniczeń instrumentu lub zakłóceń sygnału — i wyjaśnia, kiedy można je bezpiecznie zignorować, kiedy trzeba powtórzyć pomiary, a jak sensownie oszacować (imputować) brakujące dane za pomocą metod takich jak k-najbliższych sąsiadów, lasy losowe czy prosta zamiana na niską wartość. Następnie autorzy przedstawiają strategie normalizacji, które redukują niepożądaną zmienność, na przykład korygując efekty partii za pomocą próbek kontrolnych jakości lub dostosowując różnice w ilości próbki. Omawiają też transformacje, takie jak logarytmy — które łagodzą długie prawe ogony rozkładów — oraz metody skalowania, które stawiają wszystkie cząsteczki na porównywalnej skali, aby związki o dużej zmienności nie dominowały dalszych analiz. 
Testy statystyczne i wizualne opowieści
Gdy dane są właściwie przygotowane, w grę wchodzi szereg narzędzi statystycznych. Dla pojedynczych cząsteczek badacze mogą obliczać współczynniki zmian (fold change) i stosować klasyczne testy, takie jak test t lub jego nieparametryczne odpowiedniki (np. test Manna–Whitneya), by sprawdzić, czy poziomy różnią się między grupami. Przy porównaniach obejmujących kilka grup wprowadza się metody takie jak ANOVA lub test Kruskala–Wallisa, uzupełnione procedurami post hoc, które wskazują, które grupy się różnią. Skuteczność tych testów ujawnia się, gdy ich wyniki zostaną jasno zwizualizowane. Artykuł podkreśla wykresy skrzynkowe (w tym ulepszone wersje dla danych skośnych), wykresy skrzypcowe i wykresy wulkaniczne łączące wielkość efektu z istotnością statystyczną. Dla lipidów opisano też bardziej specjalistyczne wizualizacje, takie jak sieci lipidowe pokazujące skoordynowane zmiany w całych klasach oraz wykresy łańcuchów kwasów tłuszczowych ukazujące wzorce długości łańcucha węglowego i nasycenia.
Widzenie wzorców wielu zmiennych naraz
Ponieważ każda próbka może mieć setki lub tysiące zmierzonych cząsteczek, metody wielowymiarowe są kluczowe. Przegląd wyjaśnia, jak analiza głównych składowych (PCA) kondensuje tę złożoność do kilku nowych osi, które uchwytują główne kierunki zmienności, umożliwiając szybkie sprawdzenie separacji grup, efektów partii czy stabilności analizy. Bardziej zaawansowane nieliniowe metody, w tym t-SNE i UMAP, potrafią ujawnić subtelne klastry i struktury w przestrzeni wysokowymiarowej. W sytuacjach, gdy celem jest klasyfikacja próbek — na przykład odróżnienie pacjentów od kontrolnych — autorzy opisują podejścia nadzorowane oparte na metodzie najmniejszych kwadratów cząstkowych i jej ortogonalnym rozszerzeniu (PLS-DA i OPLS-DA). Metody te łączą profile molekularne z etykietami próbek, wspierają wybór cech i często są podsumowywane wykresami score, loading oraz krzywymi charakterystyki operacyjnej odbiornika (ROC).
Praktyczne zestawy narzędzi w R i Pythonie
Aby pomóc początkującym przejść od teorii do praktyki, artykuł przegląda szerokie ekosystemy pakietów. W R kolekcje takie jak tidyverse i tidymodels upraszczają obróbkę danych i modelowanie, podczas gdy ggplot2 i dodatkowe pakiety jak ggpubr, ggstatsplot i tidyplots ułatwiają tworzenie grafik gotowych do publikacji. Specjalistyczne biblioteki obsługują PCA, klasteryzację i modele oparte na PLS, a pakiety Bioconductor wspierają złożone mapy cieplne i interaktywne wykresy. W Pythonie pandas zapewnia obsługę tabel, a matplotlib, seaborn i plotly odpowiadają za wizualizację, zaś scikit-learn oferuje szeroki zestaw metod wielowymiarowych. W całym tekście autorzy kładą nacisk na przykłady krok po kroku udostępnione w towarzyszącym GitBooku, aby czytelnicy mogli odtwarzać przepływy pracy i dopasowywać je do własnych danych. 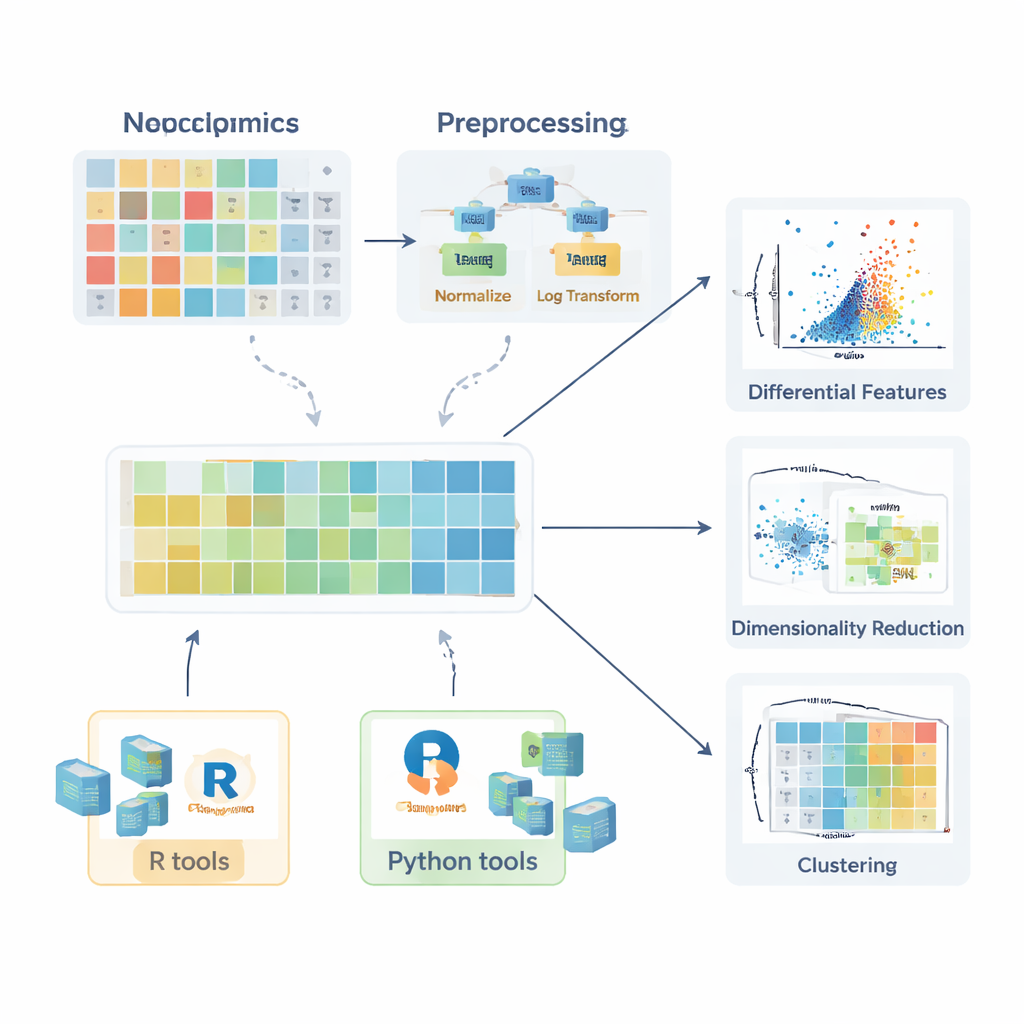
Przemiana złożonej chemii w wiarygodny wgląd
Artykuł podsumowuje, że prawdziwa obietnica lipidomiki i metabolomiki leży nie tylko w potędze instrumentów, ale w tym, jak przemyślanie przetwarza się i wizualizuje ich wyniki. Stosując dobre praktyki statystyczne, używając otwartych i dobrze udokumentowanych narzędzi w R i Pythonie oraz polegając na wspólnych przykładach kodu, badacze mogą budować solidne i powtarzalne pipeline’y. Zwiększa to szanse, że wzorce odnalezione w drobnych cząsteczkach przełożą się na wiarygodne biomarkery, lepsze zrozumienie mechanizmów chorób i bardziej spersonalizowane podejścia terapeutyczne, które ostatecznie przyniosą korzyść pacjentom.
Cytowanie: Idkowiak, J., Dehairs, J., Schwarzerová, J. et al. Best practices and tools in R and Python for statistical processing and visualization of lipidomics and metabolomics data. Nat Commun 16, 8714 (2025). https://doi.org/10.1038/s41467-025-63751-1
Słowa kluczowe: lipidomika, metabolomika, wizualizacja danych, programowanie w R, Python