Clear Sky Science · pl
Łączenie transkryptomiki przestrzennej z morfologią tkanki
Zerkając w tkanki na dwa różne sposoby
Lekarze i naukowcy coraz częściej chcą wiedzieć nie tylko które geny są aktywne w tkance, ale też dokładnie gdzie są włączane. Tymczasem mikroskopy szpitalne już rejestrują bogate obrazy struktury tkanek, z których patologowie korzystają na co dzień. Artykuł wyjaśnia, jak nowa dziedzina próbuje powiązać te dwa spojrzenia — szczegółowe mapy aktywności genów i zwykłe obrazy mikroskopowe — oraz dlaczego to połączenie może prowadzić do wcześniejszych rozpoznań, lepszego stopniowania raka i głębszego zrozumienia, jak choroby się rozwijają i rozprzestrzeniają.
Od rozproszonych komórek do map aktywności genów
Przez lata potężne metody „omikowe” wymagały rozdrabniania tkanek na mieszaninę pojedynczych komórek, co niszczyło informację o pochodzeniu każdej komórki. Transkryptomika przestrzenna to zmieniła, mierząc aktywność genów przy zachowaniu pozycji każdej komórki w tkance. Rezultatem jest siatka miejsc, z których każde ma profil ekspresji genów i precyzyjne współrzędne. Sama w sobie przestrzenna informacja o genach już ujawniła nowe wzorce różnorodności komórkowej i architektury choroby. Nie zmienia się jednak po zmierzeniu, a powtórzenie eksperymentu jest kosztowne. W przeciwieństwie do tego obrazy tkanek barwione standardowymi barwnikami, takimi jak powszechnie stosowany hematoksylina i eozyna (H&E), są tanie i powszechne oraz zawierają wizualne wskazówki dotyczące kształtu komórek, gęstości i organizacji tkanki.
Dwa sposoby łączenia obrazów i genów
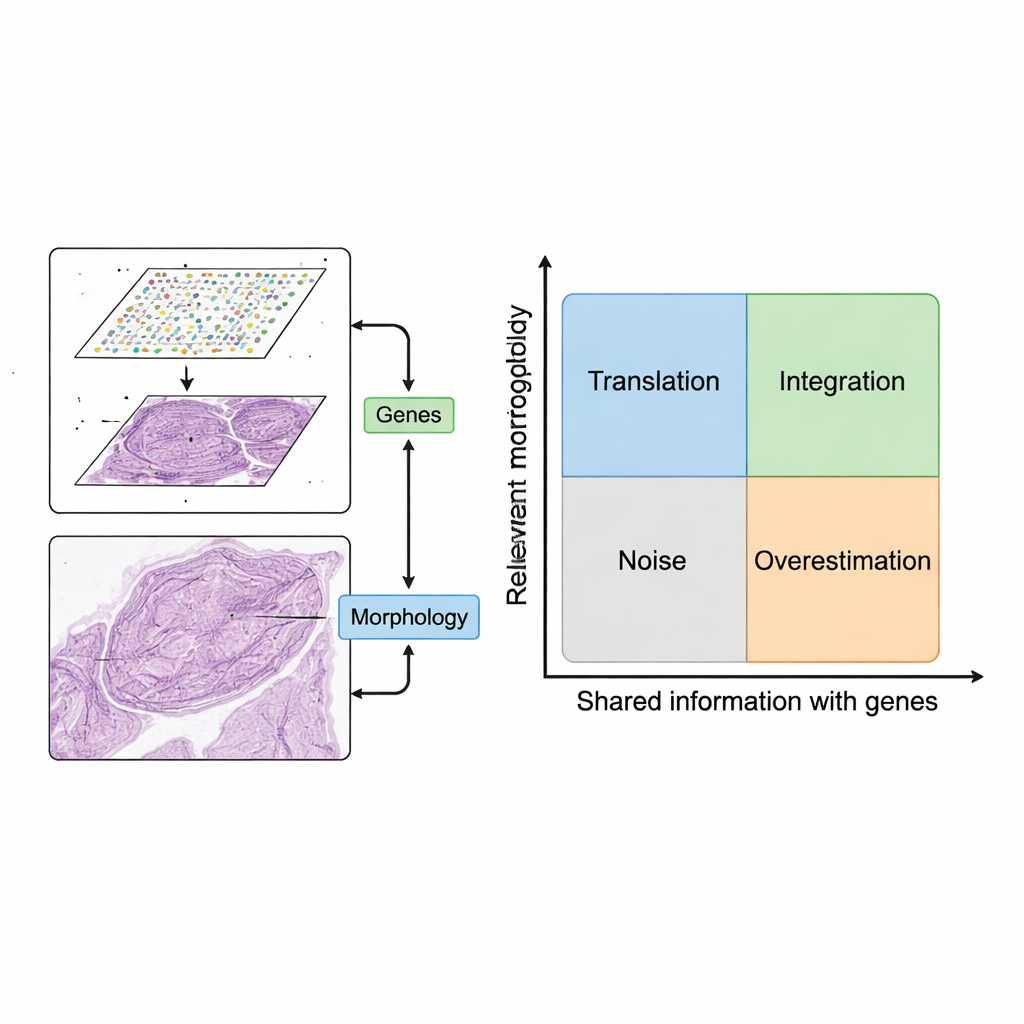
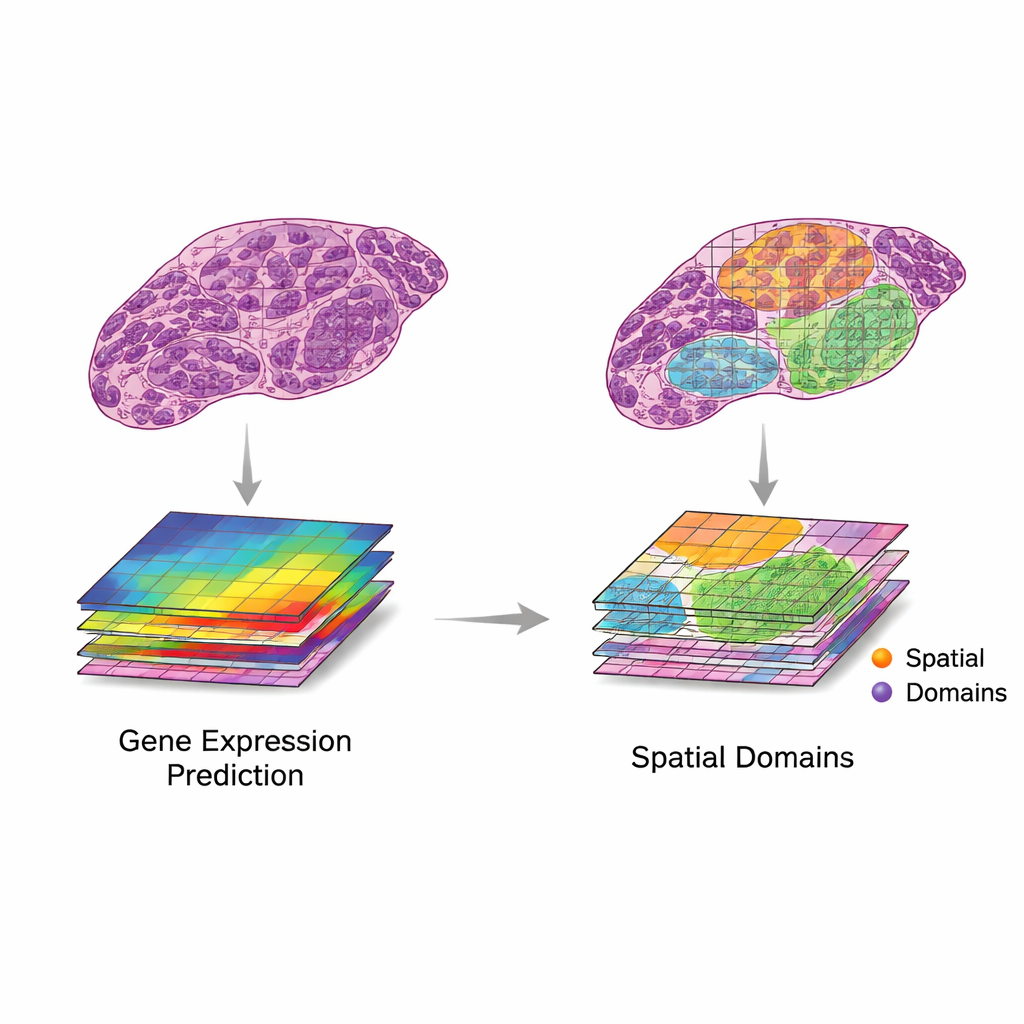
Przegląd proponuje prostą, lecz silną ramę wykorzystania tych dwóch źródeł danych razem. Najpierw fragmenty obrazu są parowane z pobliskimi punktami ekspresji genów. Potem modele komputerowe wydobywają cechy z obrazów — wzory oddające kształt, teksturę i organizację — i porównują je ze wzorcami w ekspresji genów. Autorzy opisują dwa pożądane scenariusze. W „translacji” cechy obrazu ściśle odzwierciedlają istotną aktywność genów, pozwalając modelom przewidywać, które geny są aktywne, używając tylko obrazu tkanki. Można to wykorzystać do uzupełniania brakujących pomiarów genów, osiągania lepszej rozdzielczości niż oryginalna siatka lub wnioskowania o aktywności genów na podstawie rutynowych preparatów klinicznych bez dodatkowych badań laboratoryjnych. W „integracji” cechy obrazu uchwycą użyteczną informację, którą dane genetyczne pomijają, na przykład powolne zmiany strukturalne lub subtelną organizację tkanki, pomagając wyznaczyć wyraźniejsze regiony lub „domeny” wewnątrz tkanki.
Kiedy dodatkowa informacja pomaga — a kiedy szkodzi
Nie każda cecha obrazu jest warta użycia. Autorzy wprowadzają mapę pojęciową z dwiema osiami: jak istotna jest cecha obrazu dla pytania biologicznego oraz jak bardzo pokrywa się z informacją genetyczną. Cechy, które nie są ani istotne, ani powiązane z genami, stanowią szum, takie jak artefakty barwienia. Cechy, które śledzą wzorce genów, ale odnoszą się do nieistotnych genów (np. podstawowych genów „housekeeping”), mogą sprawiać, że modele wyglądają dobrze w testach, a jednocześnie wnoszą niewielką wartość kliniczną. Organizując metody w czterech ćwiartkach — translacja, integracja, szum i przeszacowanie — rama wyjaśnia, kiedy łączenie obrazów i genów rzeczywiście dodaje wgląd, a kiedy po prostu powtarza lub zaciera to, co już wiadomo.
Obecne narzędzia, testy i rosnące trudności
Szybko rozwijająca się fala metod sztucznej inteligencji próbuje obecnie realizować translację i integrację na rzeczywistych danych. Wczesne systemy opierały się na splotowych sieciach neuronowych, podczas gdy nowsze wykorzystują transformatory, grafowe sieci neuronowe i modele wieloskalowe, które potrafią przetwarzać szczegóły od maleńkich struktur komórkowych po kontekst całego preparatu. Metody te stosowano do przewidywania aktywności genów z obrazów H&E, generowania map o superrozdzielczości oraz pomagania w identyfikacji regionów tkanki o odrębnym zachowaniu. Do oceny wydajności badacze polegają na miarach statystycznych, takich jak korelacja między przewidywanymi a zaobserwowanymi poziomami genów czy zgodność między regionami zdefiniowanymi przez AI a oznaczeniami ekspertów-patologów. Jednak zbiory danych są wciąż małe i zróżnicowane, a porównania między studiami są trudne. Wiele zgłaszanych osiągnięć może odzwierciedlać przeuczenie albo sukces w odniesieniu do genów i wzorców, które mają niewielkie znaczenie kliniczne.
Dokąd to może prowadzić
Autorzy konkludują, że łączenie przestrzennych map genów z obrazami tkanek to obiecujące, lecz wciąż wczesne przedsięwzięcie. Dzisiejsze modele często osiągają jedynie umiarkowaną dokładność i nie są jeszcze gotowe do rutynowego zastosowania medycznego. Przyszły postęp prawdopodobnie wynikać będzie z lepszych cech obrazu, zwłaszcza dużych „modeli fundamentowych” trenowanych na milionach preparatów patologicznych, oraz z koncentrowania się na genach i wzorcach, które rzeczywiście wpływają na opiekę nad pacjentem. Starannie zaprojektowana integracja pewnego dnia może ujawnić wczesne sygnały ostrzegawcze choroby poprzez wykrywanie rozbieżności między tym, jak tkanka wygląda teraz, a tym, co jej geny przewidują, że wydarzy się później. Krótko mówiąc, ta praca wyznacza mapę drogową przekształcania rutynowych obrazów mikroskopowych w bogate, genetycznie informowane mapy, które pomogą lekarzom zrozumieć i leczyć choroby precyzyjniej.
Cytowanie: Chelebian, E., Avenel, C. & Wählby, C. Combining spatial transcriptomics with tissue morphology. Nat Commun 16, 4452 (2025). https://doi.org/10.1038/s41467-025-58989-8
Słowa kluczowe: transkryptomika przestrzenna, morfologia tkanki, patologia cyfrowa, predykcja ekspresji genów, AI obrazowa