Clear Sky Science · pl
Aktywacja kaspaz wzmacnia przeciw-Desmogleinie 3 indukowaną akantolizę w ludzkiej naskórku
Gdy organizm atakuje własny „klej” skórny
Pemfigus zwykły to rzadka, lecz groźna choroba, w której układ odpornościowy atakuje „klej” utrzymujący komórki skóry razem, powodując bolesne pęcherze i otwarte rany. W tym badaniu przyjrzano się sprawie pod mikroskopem, by odpowiedzieć na kluczowe pytanie: czy te pęcherze powstają wyłącznie dlatego, że przeciwciała blokują ten klej, czy też dodatkowe sygnały związane ze śmiercią komórki pomagają rozdzierać komórki skóry? Zrozumienie tego może prowadzić do bardziej ukierunkowanych i łagodniejszych terapii dla pacjentów.
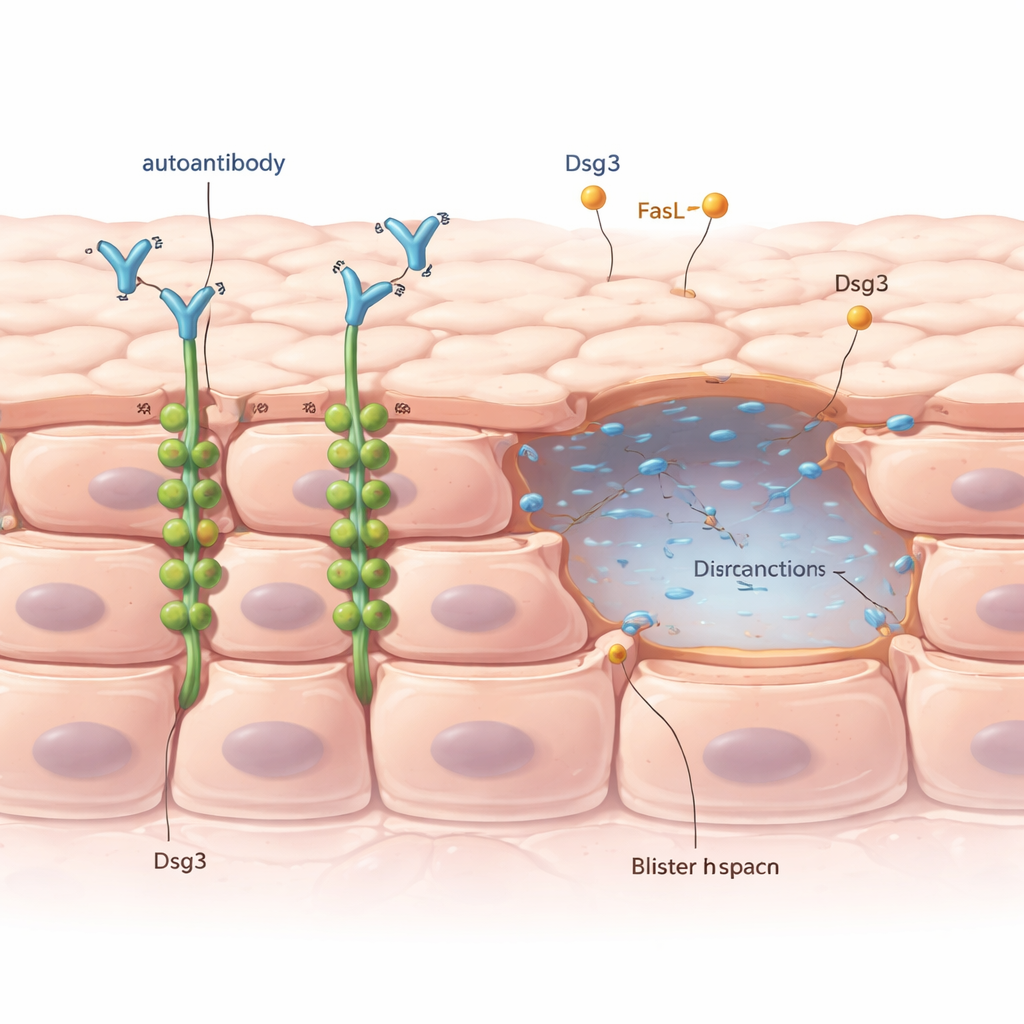
Jak komórki skóry normalnie pozostają zwarte
Zewnętrzna warstwa skóry zbudowana jest z ciasno upakowanych komórek połączonych przez maleńkie struktury zwane desmosomami, które działają jak nitowania między sąsiednimi komórkami. Kluczowym składnikiem tych „nitów” jest białko zwane desmogleiną 3, w skrócie Dsg3. W pemfigusie zwykłym pacjenci wytwarzają przeciwciała, które omyłkowo wiążą się z Dsg3. Lekarze od dawna wiedzą, że ten atak przeciwciał prowadzi do procesu zwanego akantolizą, w którym komórki skóry tracą kontakt i pojawiają się pęcherze. Tajemnicą pozostaje jednak to, dlaczego pęcherze występują tylko w niektórych miejscach i w pewnych momentach, mimo że szkodliwe przeciwciała rozprzestrzeniają się po całej skórze.
Samo obecność przeciwciał może rozpocząć uszkodzenie
Aby sprawdzić, co robią same przeciwciała, badacze użyli fragmentów zdrowej ludzkiej skóry oraz hodowanej w laboratorium linii komórek skórnych. Ekspozycja na dobrze poznane przeciwciało przeciw Dsg3 osłabiła adhezję międzykomórkową i wywołała akantolizę nawet bez uruchamiania typowych mechanizmów śmierci komórki wewnątrz komórek. Kluczowym zdarzeniem było odciągnięcie Dsg3 z powierzchni komórki do jej wnętrza, często zapakowanego w drobne pęcherzyki. Usunięcie Dsg3 z „nitów” między komórkami skracało i osłabiało desmosomy, pozwalając komórkom zacząć się rozdzielać.
Gdy dołączają sygnały śmierci komórki, jest gorzej
Zespół zadał następnie pytanie, czy znany w pemfigusie sygnał śmierci komórki — cząsteczka zwana ligandom Fas (FasL) — zmienia ten obraz. FasL może aktywować enzymy zwane kaspazami, w szczególności kaspazę-8, które zwykle kierują komórki ku zaprogramowanej śmierci. W próbkach skóry od pacjentów i w modelu skórnym badacze zaobserwowali aktywną kaspazę-8 w obszarach z uszkodzeniem, ale bez klasycznych oznak umierających komórek. Gdy dodano niewielkie, nieśmiertelne ilości FasL razem z przeciwciałem przeciw Dsg3, pęcherze powstawały szybciej i bardziej intensywnie, szczególnie w głębszych warstwach naskórka. W hodowli komórkowej sam FasL nie osłabiał adhezji, ale w połączeniu z przeciwciałem znacznie zwiększał rozpad komórek — a ten efekt znikał po zablokowaniu kaspazy-8.
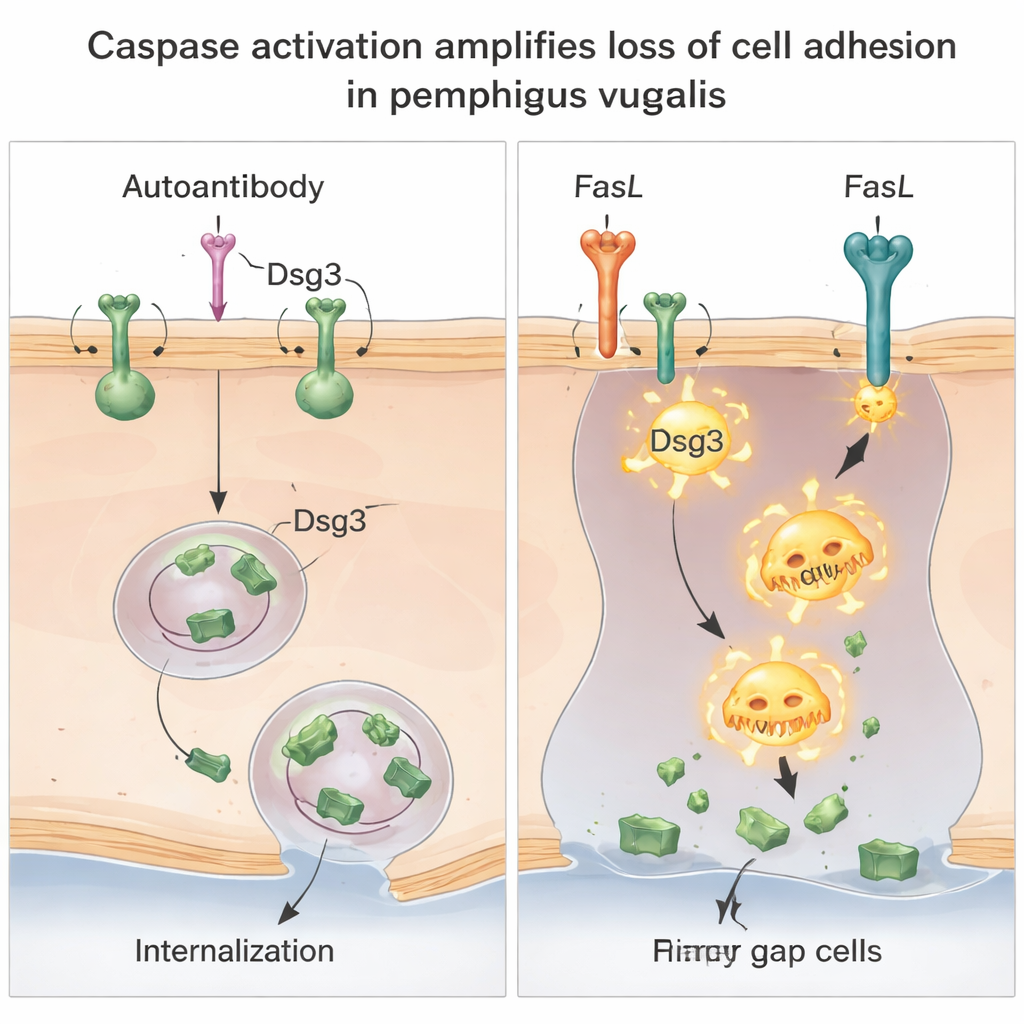
Dwie drogi do zerwania tej samej spoiny
Wnikliwsze badania pokazały, że istnieją w rzeczywistości dwie odrębne, lecz współdziałające drogi utraty Dsg3. Po pierwsze, przeciwciało sprzyja internalizacji intactnych kompleksów Dsg3–przeciwciało z powierzchni komórki do wyspecjalizowanych pęcherzyków błonowych, zmniejszając ilość Dsg3 dostępnego w połączeniach komórkowych. Ten etap nie zależał od kaspaz i nie był zatrzymywany przez blokowanie powszechnych szlaków degradacji białek, co sugeruje specyficzne przemeblowanie Dsg3 wewnątrz komórki. Po drugie, gdy obecny jest FasL, kaspaza-8 staje się aktywna i tnie Dsg3 na mniejsze fragmenty, szczególnie w luźniej przylegających częściach błony. To cięcie zależne od kaspazy jeszcze bardziej zmniejsza ilość pełnej, funkcjonalnej Dsg3 i potęguje utratę adhezji wywołaną przez przeciwciała.
Co to znaczy dla pacjentów i przyszłych terapii
Podsumowując, wyniki sugerują, że w pemfigusie zwykłym przeciwciała przeciw Dsg3 rozpoczynają proces powstawania pęcherzy poprzez usuwanie Dsg3 z powierzchni komórki, natomiast aktywacja kaspaz napędzana przez FasL działa jak wzmacniacz, który znacząco pogłębia uszkodzenie poprzez przecinanie pozostałej Dsg3. Co ważne, wiele z tych procesów zachodzi zanim komórki skóry faktycznie umrą. Dla pacjentów oznacza to, że skuteczne terapie mogą nie tylko redukować szkodliwe przeciwciała, lecz także blokować FasL lub kaspazy, aby zapobiegać tworzeniu się lub rozszerzaniu pęcherzy. Ten dualny mechanizm może wyjaśnić, dlaczego choroba może wyglądać i zachowywać się tak różnie u różnych osób — lub w różnych miejscach na ciele — i wskazuje na nowe, bardziej precyzyjne sposoby zachowania naturalnego „kleju” skóry.
Cytowanie: Schmidt, M.F., Feoktistova, M.A., Panayotova-Dimitrova, D. et al. Caspase-activation powers anti-Desmoglein 3-induced acantholysis in human epidermis. Cell Death Discov. 12, 102 (2026). https://doi.org/10.1038/s41420-026-02963-w
Słowa kluczowe: pemfigus zwykły, autoimmunologiczne pęcherzowe choroby skóry, desmogleina 3, kaspaza-8, ligand Fas