Clear Sky Science · pl
Modyfikowanie sygnatur metabolicznych w celu ograniczenia oporności na kabozantynib w modelach komórkowych ostrej białaczki szpikowej z FLT3‑ITD
Dlaczego ma to znaczenie dla leczenia raka
Wiele nowoczesnych leków przeciwnowotworowych jest zaprojektowanych tak, by atakować pojedyncze wadliwe białko w komórkach guza. Leki celowane potrafią wywołać spektakularne remisje, ale nowotwory często znajdują sposoby, by się zaadaptować i odrosnąć. Artykuł analizuje, jak pewien rodzaj nowotworu krwi — ostra białaczka szpikowa (AML) — rozwija oporność na jeden z takich leków celowanych, kabozantynib, oraz jak przeprogramowanie wykorzystania energii przez komórki nowotworowe może pomóc lekarzom przechytrzyć tę oporność.
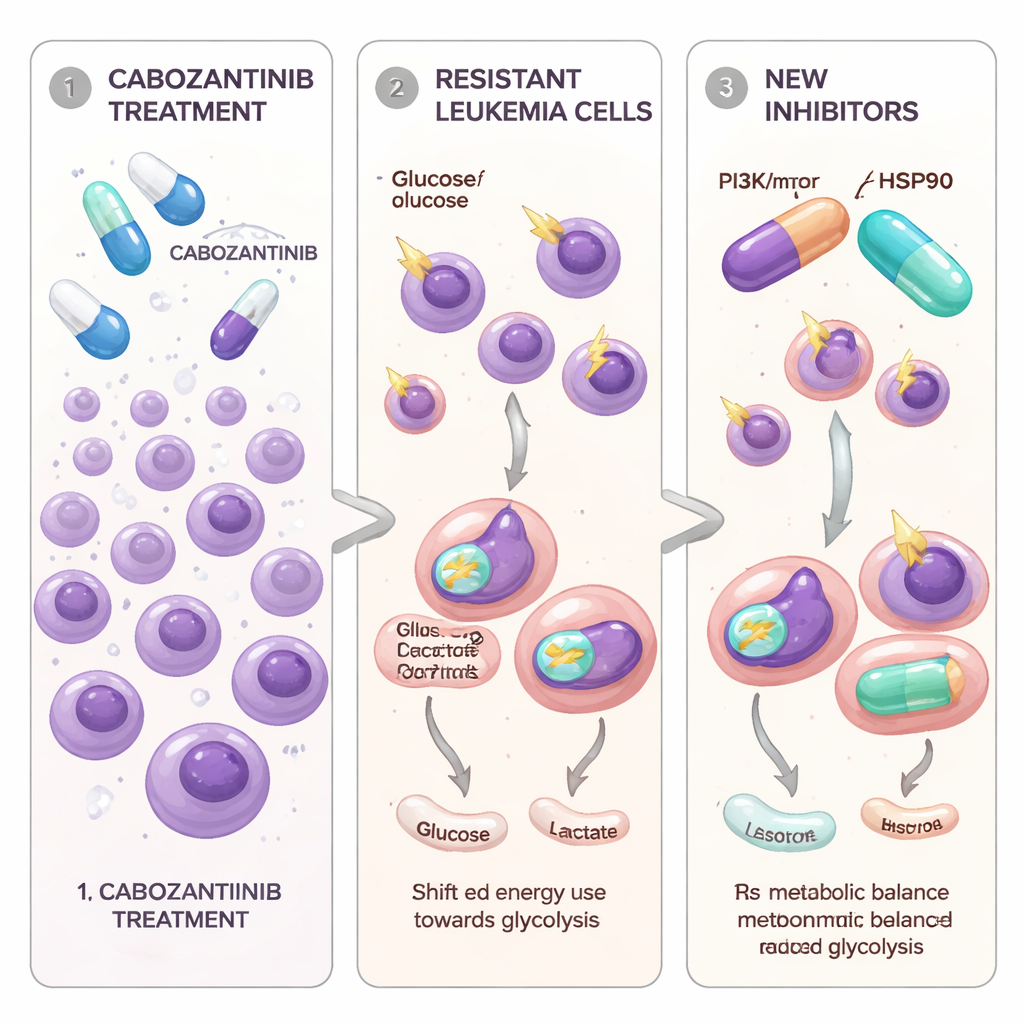
Komórki białaczki, które uczą się unikać leku celowanego
Naukowcy skoncentrowali się na komórkach AML z mutacją w przełączniku sygnałowym zwanym FLT3‑ITD, która napędza szczególnie agresywną postać choroby. Kabozantynib, doustny lek stosowany już w kilku nowotworach litych, potrafi silnie blokować komórki białaczki zależne od FLT3 w warunkach laboratoryjnych. Aby odwzorować to, co dzieje się u pacjentów w czasie, zespół stopniowo wystawiał dwie linie komórkowe z mutacją FLT3 na rosnące dawki kabozantynibu, aż niektóre komórki przeżyły i zaczęły się ponownie namnażać. Nowe populacje komórek, nazwane Molm13‑XR i MV4‑11‑XR, tolerowały stężenia kabozantynibu wielokrotnie wyższe niż ich „rodzicielskie” komórki. Stały się także mniej wrażliwe na dwa inne zatwierdzone leki skierowane przeciwko FLT3, sorafenib i quizartinib, przy jednoczesnym zachowaniu wrażliwości na inny inhibitor, gilterytynib.
Genetyczne modyfikacje wspierające przeżycie raka
Analiza molekularna wykazała, że komórki oporne na lek niosły nowe zmiany w genie FLT3. Obie linie oporne nabyły tę samą mutację punktową, nazwaną D835Y, w kluczowym obszarze domeny kinazy FLT3 — znanym gorącym punkcie oporności na kilka leków. Linia MV4‑11‑XR dodatkowo zyskała nietypowe usunięcie o długości około 1,3 kb, które wyeliminowało cały ekson FLT3, kasując część domeny ważnej dla wiązania leku. Zmiany te wydają się być wynikiem selekcji podczas długotrwałej ekspozycji na kabozantynib: zmutowane formy FLT3 stały się znacznie częstsze w komórkach opornych niż w populacji wyjściowej. Równocześnie kluczowe szlaki sygnałowe położone poniżej FLT3 — takie jak ERK, STAT5 i AKT — były silniej włączone, wspierając szybszy wzrost i większą formację kolonii w komórkach opornych.
Komórki raka zmieniają systemy zasilania
Zespół zapytał następnie, czy oporność związana jest nie tylko z genetyką, lecz także ze sposobem pozyskiwania energii przez komórki. Dzięki sekwencjonowaniu RNA i testom metabolicznym odkryto spójny wzorzec: komórki oporne na kabozantynib w znacznie większym stopniu polegały na glikolizie — szybkim rozpadowi glukozy w płynie komórkowym — nawet przy wystarczającej ilości tlenu. Komórki te pobierały więcej glukozy, produkowały więcej mleczanu, wykazywały wyższą aktywność kluczowego enzymu GAPDH i zwiększoną ekspresję wielu genów związanych z glikolizą. Natomiast mitochondria, struktury odpowiadające za bardziej wydajne wytwarzanie energii, były mniej aktywne i mniej liczne. Pomiary wykorzystania tlenu wykazały obniżenie zarówno podstawowego, jak i maksymalnego oddechu mitochondrialnego, a wewnątrzkomórkowe rodniki tlenowe były podwyższone, co wskazuje na zestresowane, niedomagające mitochondria.
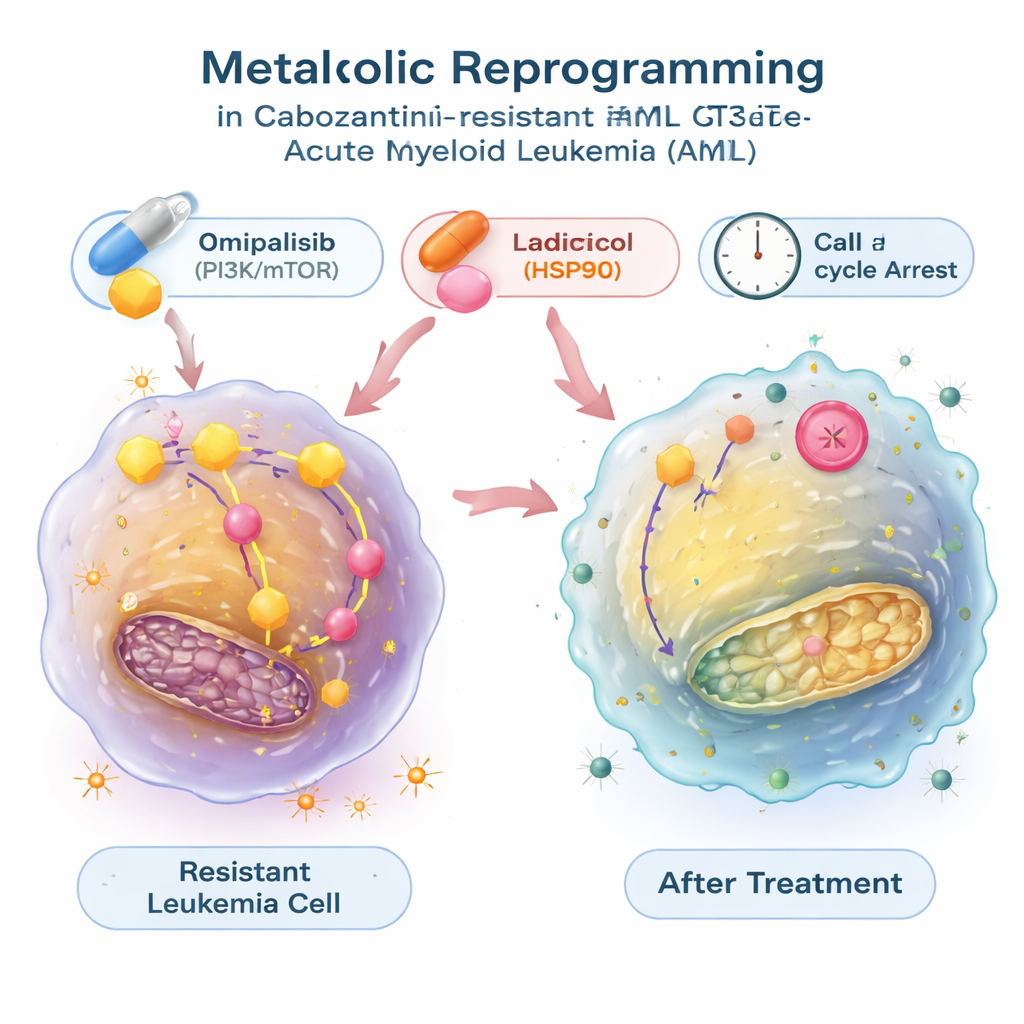
Wyszukiwanie leków, które odwrócą przełączenie metaboliczne
Aby sprawdzić, czy można cofnąć tę zmianę energetyczną, badacze skorzystali z dużej publicznej bazy danych łączącej wzorce ekspresji genów z efektami tysięcy związków. Szukali leków przewidywanych jako przeciwdziałające sygnaturze metabolicznej komórek opornych i wytypowali dwa: radicicol, który blokuje białkowego czaperona HSP90, oraz omipalisib, inhibitor szlaku PI3K/mTOR kontrolującego wzrost i metabolizm. W testach laboratoryjnych obie molekuły nie tylko spowalniały wzrost komórek opornych, ale też redukowały ich nadaktywną glikolizę — normalizowały pobór glukozy i wydzielanie mleczanu oraz obniżały ekspresję genów związanych z glikolizą. Związki te wprowadzały komórki białaczki w fazę spoczynkową cyklu komórkowego, a w przypadku radicicolu dodatkowo indukowały znaczną apoptozę. W połączeniu z kabozantynibem omipalisib — a w jednym modelu także radicicol — działały synergistycznie, ułatwiając eliminację komórek opornych.
Co to oznacza dla przyszłych terapii białaczki
Dla osób niezajmujących się specjalistycznie tematem przesłanie jest takie: komórki białaczki mogą uniknąć leku celowanego nie tylko przez mutację jego bezpośredniego celu, lecz także przez zmianę sposobu, w jaki wytwarzają i wykorzystują energię. Badanie pokazuje, że komórki AML oporne na kabozantynib przyjmują strategię „spalania cukru”, jednocześnie zaniedbując mitochondria. Uderzając w szlaki wspierające to przeprogramowanie metabolizmu — za pomocą leków takich jak omipalisib czy inhibitory HSP90 — może być możliwe przywrócenie wrażliwości na kabozantynib i podobne terapie. Choć wnioski pochodzą z modeli komórkowych, a nie od pacjentów, sugerują, że łączenie leków celowanych z substancjami modulującymi metabolizm może być obiecującym sposobem opóźniania lub przezwyciężania oporności w AML z mutacją FLT3.
Cytowanie: Fu, YH., Ng, K.M., Tseng, CY. et al. Modulating metabolic signatures to mitigate cabozantinib resistance in FLT3-ITD acute myeloid leukemia cell models. Cell Death Discov. 12, 98 (2026). https://doi.org/10.1038/s41420-026-02957-8
Słowa kluczowe: ostra białaczka szpikowa, oporność na leki, mutacja FLT3, metabolizm nowotworu, kabozantynib