Clear Sky Science · pl
Hamowanie RRM1 uczula gruczolakoraka płuca na leczenie deksacytybiną
Przemiana letniego leku w silniejszego sojusznika
Rak płuca pozostaje jednym z najgroźniejszych nowotworów, a wielu pacjentów ostatecznie wyczerpuje dostępne skuteczne opcje terapeutyczne. Lekarze od dawna liczyli na to, że leki, które delikatnie przeprogramowują DNA komórek nowotworowych zamiast po prostu zatruwać dzielące się komórki, mogą pomóc. Jednym z takich leków jest deksacytybina, która sprawdza się w nowotworach krwi, ale zawiodła w guzach litych, takich jak rak płuca. W badaniu postawiono proste, praktyczne pytanie o dalekosiężnych implikacjach: czy da się sprawić, by guzy płuc w końcu zareagowały na deksacytybinę, wykorzystując narzędzia, które już dobrze znamy?
Dlaczego sprawdzony lek zawodzi w guzach litych
Deksacytybina jest analogiem jednego z budulców DNA. Gdy wślizguje się do DNA komórki podczas jego kopiowania, może usuwać nieprawidłowe chemiczne znaczniki, które wyciszają geny ochronne, w tym supresory nowotworowe i geny układu odpornościowego. W białaczkach pomaga to przywrócić komórki do zdrowszego stanu. W guzach płuc jednak lek działa słabo. Autorzy przypuszczali, że problem nie leży w mechanizmie działania deksacytybiny, lecz w tym, jak niewiele z niej trafia rzeczywiście do DNA komórek guza. Mierząc maleńkie ilości leku wbudowanego w DNA w wielu liniach komórkowych, potwierdzili, że komórki, które wchłaniały więcej deksacytybiny, były znacznie łatwiejsze do zabicia tym lekiem.
Komórkowy strażnik, który blokuje lek
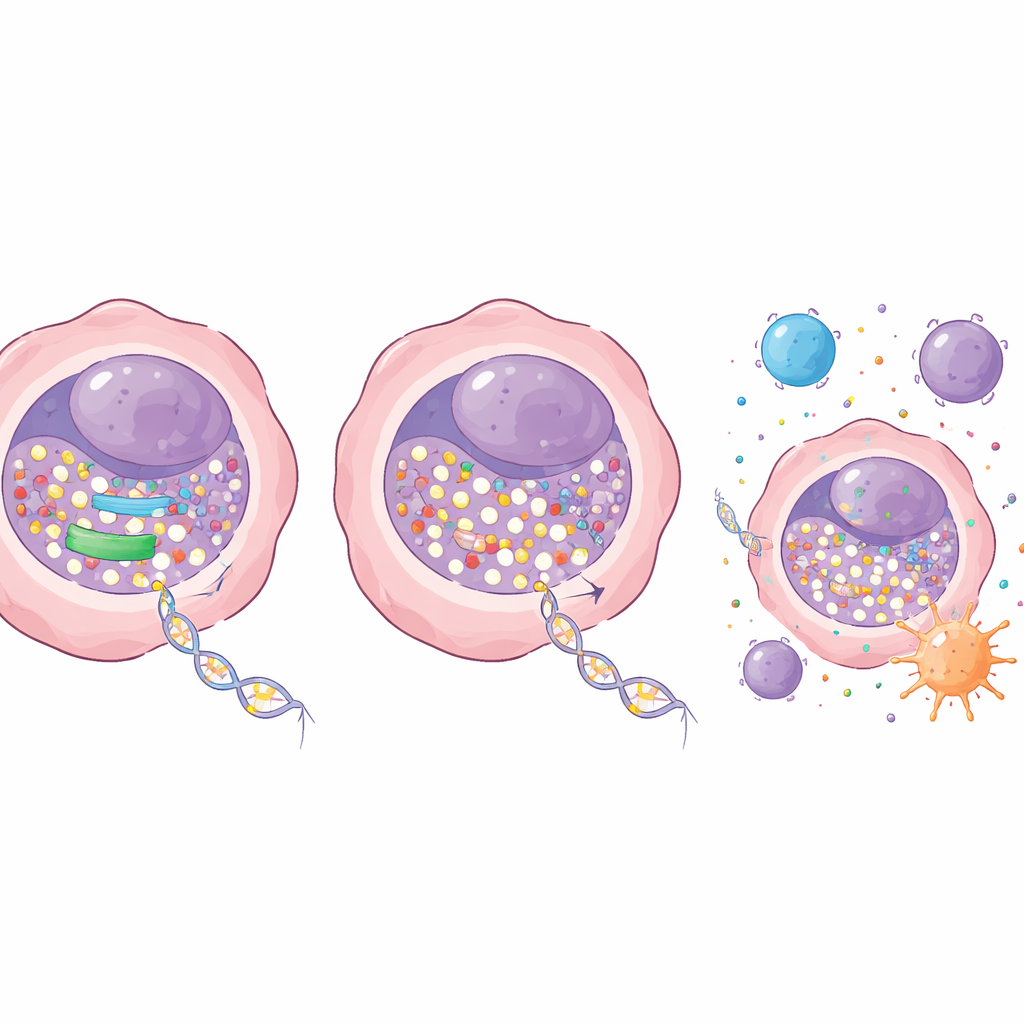
Aby ustalić, co ogranicza wejście leku do DNA, badacze zbadali geny zaangażowane w przetwarzanie nukleozydów — surowców do budowy DNA. Na wyróżnienie zasługiwał jeden enzym, zwany RRM1. RRM1 jest częścią kompleksu przekształcającego zwykłe podjednostki w aktywne formy wykorzystywane do syntezy DNA. W gruczolakorakach płuca enzym ten był wyjątkowo obficie obecny w guzach w porównaniu z prawidłową tkanką płucną, a pacjenci z niższym poziomem RRM1 mieli tendencję do dłuższego przeżycia. W panelu linii nowotworowych wyższe poziomy RRM1 szły w parze z mniejszą inkorporacją deksacytybiny, co silnie sugeruje, że enzym ten działa jako strażnik, wypierający lek.
Rozbrojenie strażnika, by pomóc lekowi
Zespół zapytał następnie, co się stanie, jeśli częściowo wyłączy się RRM1. Za pomocą narzędzi genetycznych zmniejszyli ekspresję RRM1 w komórkach raka płuca bez zabijania ich bezpośrednio. Sama redukcja miała tylko łagodny wpływ na wzrost. Ale w połączeniu z niskimi dawkami deksacytybiny efekt był dramatyczny: kolonie komórek raka płuca silnie się kurczyły w hodowlach, a guzy rosły znacznie wolniej u myszy. Co ważne, skuteczne dawki były dobrze tolerowane — nie obserwowano oczywistych uszkodzeń krwi, wątroby ani nerek u zwierząt. Na poziomie molekularnym blokada RRM1 pozwoliła na większą inkorporację deksacytybiny do DNA, co prowadziło do silniejszej utraty enzymu dodającego metylacje, DNMT1, i większego spadku globalnej metylacji DNA. To z kolei przywracało do działania wyciszone geny supresorowe.
Włączenie alarmu immunologicznego w obrębie guzów
Pozostawiając na boku spowolnienie podziałów, terapia skojarzona zmieniła sposób, w jaki komórki nowotworowe wchodzą w interakcje z układem odpornościowym. Większa ilość deksacytybiny w DNA nasiliła sygnały uszkodzenia DNA wewnątrz komórek i skłoniła je ku zaprogramowanej śmierci. Jednocześnie zwiększyła aktywność wewnętrznego systemu alarmowego skupionego wokół szlaku STING, który wykrywa błędnie umieszczone DNA i uruchamia odpowiedzi przypominające reakcję przeciwwirusową. Gdy RRM1 był zablokowany, deksacytybina silniej aktywowała ten szlak i jego geny docelowe, w tym te, które przyciągają i pobudzają komórki odpornościowe. W modelach mysich z zachowanym układem odpornościowym łączenie deksacytybiny z inhibitorem RRM1 dawało silniejszą kontrolę guza niż każde z leczeń osobno, bez wyraźnego wzrostu toksyczności. Autorzy wykazali też, że strategia blokowania tego enzymu wzmacnia działanie specyficznie deksacytybiny, a w przypadku spokrewnionego leku azacytydyny może nawet działać przeciwnie, co podkreśla potrzebę dobrania właściwych partnerów.
Co to może znaczyć dla pacjentów
W całości praca maluje jasny obraz: nadaktywność enzymu budującego DNA w guzach płuc ogranicza ilość deksacytybiny trafiającej do miejsca działania. Częściowo blokując ten enzym, zmuszamy komórki nowotworowe do wykorzystania więcej leku zamiast zwykłych podjednostek. Ta zmiana pozwala niskim dawkom deksacytybiny skuteczniej przywracać aktywność genów ochronnych, uszkadzać DNA komórek nowotworowych i budzić mechanizmy odpornościowe, zachowując przy tym akceptowalną tolerancję w modelach zwierzęcych. Dla pacjentów sugeruje to realistyczną drogę: ponowne zastosowanie lub udoskonalenie inhibitorów RRM1 w skojarzeniu z niskimi dawkami deksacytybiny i potencjalnie z nowoczesnymi immunoterapiami, aby przemienić niegdyś rozczarowujący lek w użyteczny element terapii raka płuca.
Cytowanie: Jiang, N., Liu, J., Vaghasia, A. et al. RRM1 inhibition sensitizes lung adenocarcinoma to decitabine treatment. Cell Death Dis 17, 275 (2026). https://doi.org/10.1038/s41419-026-08522-6
Słowa kluczowe: gruczolakorak płuca, deksacytybina, metylacja DNA, reduktaza rybonukleotydowa, immunoterapia przeciwnowotworowa