Clear Sky Science · pl
PGC-1α chroni przed MASH poprzez zależną od Tim23 hamującą aktywność DRP1 prowadzącą do ferroptozy
Dlaczego to ma znaczenie dla zdrowia codziennego
U wielu osób z otyłością lub cukrzycą typu 2 cicho rozwija się poważny problem wątroby zwany metabolicznie związanym zapaleniem ze stłuszczeniem wątroby (MASH). W tym stanie komórki wątroby przepełnione tłuszczem ulegają zapaleniu i zaczynają obumierać, co otwiera drogę do bliznowacenia, marskości i raka wątroby. Badanie odkrywa ukryty system samoochrony wewnątrz komórek wątroby — skupiony wokół maleńkich elektrowni komórkowych, czyli mitochondriów — który może albo chronić wątrobę przed uszkodzeniem, albo w razie jego zawiedzenia przyspieszać chorobę. Zrozumienie tej wewnętrznej blokady bezpieczeństwa może otworzyć drzwi do nowych terapii przeciw jednemu z najpowszechniejszych zagrożeń dla wątroby na świecie.
Bliższe spojrzenie na cichą chorobę wątroby
MASH rozwija się, gdy proste stłuszczenie wątroby przechyla się w bardziej niebezpieczny stan charakteryzujący się obrzękiem i uszkodzeniem komórek wątroby, zapaleniem i w końcu tkanką bliznowatą. Autorzy zbadali próbki wątroby od pacjentów z MASH oraz od myszy karmionych dietami wysokotłuszczowymi, bogatymi w cukry lub ubogimi w składniki odżywcze, które naśladują stan ludzki. Skupili się na szczególnym typie śmierci komórki zwanej ferroptozą, w której żelazo i uszkodzone tłuszcze łączą się, tworząc toksyczne cząsteczki, które powodują ubytki w błonach komórkowych. Zarówno u ludzi, jak i u myszy z MASH komórki wątroby wykazywały cechy tej żelazowo‑i lipidowo napędzanej śmierci: nadmiar depozytów żelaza, zdeformowane mitochondria oraz wysokie poziomy białek sprzyjających uszkodzeniom lipidów, przy jednocześnie niskich poziomach białek, które normalnie detoksyfikują szkodliwe produkty uboczne.
Dowody, że blokowanie żelazowo‑napędzanej śmierci pomaga
Aby sprawdzić, czy ferroptoza jest jedynie towarzyszem czy napędem choroby, badacze leczyli myszy na diecie wysokotłuszczowej ferrostatyną‑1, związkiem specyficznie blokującym ferroptozę. Myszy otrzymujące inhibitor miały mniej nagromadzonego tłuszczu, mniejsze przeciążenie żelazem i mniej objawów zapalenia oraz bliznowacenia w wątrobie. Badania krwi wykazały poprawę funkcji wątroby i lepsze parametry metaboliczne, w tym niższy cholesterol i lepszą wrażliwość na insulinę. W izolowanych komórkach wątroby myszy wystawionych na kwas palmitynowy — tłuszcz naśladujący przeciążenie widoczne w MASH — ten sam lek zmniejszył gromadzenie tłuszczu, ładowanie żelazem, uszkodzenia oksydacyjne oraz sygnały zapalne. Razem te wyniki przemawiają za tym, że ferroptoza jest kluczowym silnikiem szkód w MASH i że przerwanie tego procesu może istotnie złagodzić przebieg choroby.
Wbudowany obrońca w mitochondriach wątroby
Zespół skupił się następnie na PGC‑1α, głównym regulatorze, który pomaga mitochondriom wytwarzać energię i radzić sobie ze stresem. W wątrobie ludzi z MASH, podobnie jak u chorych myszy i zestresowanych komórek wątroby, poziomy PGC‑1α były wyraźnie obniżone, podczas gdy białko rozszczepiające mitochondria DRP1 i enzym aktywujący lipidy ACSL4 były podwyższone. U mysi z inżynierią genetyczną pozbawionych PGC‑1α jedynie w komórkach wątroby autorzy stwierdzili, że utrata tego obrońcy uczyniła diety wysokotłuszczowe znacznie bardziej szkodliwymi: wątroby były bardziej stłuszczone, bardziej zapalne, bardziej przeciążone żelazem i wykazywały silniejsze sygnały ferroptozy. Na poziomie komórkowym niedobór PGC‑1α zwiększał aktywność DRP1, podnosił poziomy ACSL4 i białek importu żelaza oraz osłabiał mechanizmy antyoksydacyjne, które normalnie hamują ferroptozę.
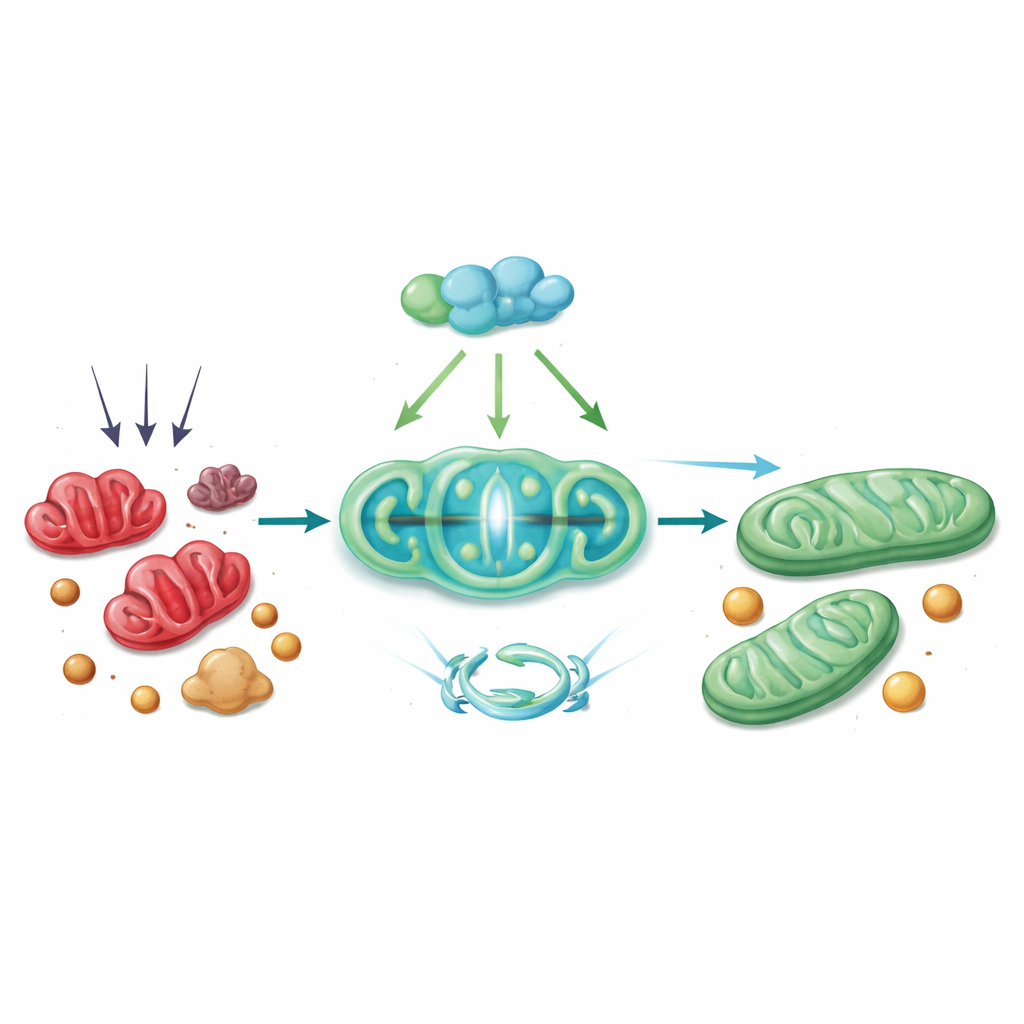
Ochronny łańcuch reakcji wewnątrz komórki
Mechanistycznie PGC‑1α działa przez łańcuch partnerów. Współpracuje z czynnikiem transkrypcyjnym Nrf1, aby zwiększyć produkcję Tim23, kanału w wewnętrznej błonie mitochondrialnej niezbędnego do importu białek i utrzymania zdrowej struktury. Gdy poziomy Tim23 spadają, potencjał błony mitochondrialnej słabnie, co wyzwala DRP1 do fragmentacji organelli. Badanie pokazuje, że przy zmniejszonym Tim23 DRP1 jest bardziej aktywny i częściej współdziała z ACSL4 na powierzchni mitochondriów, przyciągając ten enzym modyfikujący lipidy do mitochondriów. Tam ACSL4 pomaga zaszczepić zmiany lipidowe, które czynią komórki podatnymi na ferroptozę. Przywrócenie PGC‑1α — zarówno u myszy za pomocą wektora wirusowego dostarczającego gen, jak i w hodowlach hepatocytów przy użyciu aktywatora opartego na CRISPR — odwróciło wiele z tych kroków: Tim23 wzrósł, aktywność DRP1 i ACSL4 spadła, mitochondria wyglądały zdrowiej, a markery ferroptozy i uszkodzenia wątroby zmalały.
Jak to odkrycie może ukierunkować przyszłe terapie
Dla osoby niebędącej specjalistą główny wniosek jest taki, że wątroba posiada wewnętrzny hamulec przeciw żelazowo‑i tłuszczowo napędzanej śmierci komórek, a ten hamulec jest wbudowany w mitochondria. Łańcuch PGC‑1α–Tim23–DRP1–ACSL4 działa jak obwód bezpieczeństwa: gdy PGC‑1α jest silny, Tim23 utrzymuje stabilność mitochondriów, DRP1 i ACSL4 są powściągliwe, a komórki wątroby są mniej skłonne do samozniszczenia. Gdy ten obwód zawiedzie, ferroptoza przyspiesza, a MASH się pogarsza. Identyfikując tę ścieżkę w tkance ludzkiej i modelach zwierzęcych, badanie wskazuje dwa komplementarne kierunki przyszłych terapii — bezpośrednie blokowanie ferroptozy oraz zwiększanie aktywności PGC‑1α lub Tim23 w celu stabilizacji mitochondriów — co daje nadzieję na wcześniejsze i skuteczniejsze interwencje, zanim dojdzie do nieodwracalnego bliznowacenia wątroby.
Cytowanie: Zhao, Y., Zhang, L., Li, B. et al. PGC-1α protects against MASH via Tim23-dependent inhibition of DRP1-mediated ferroptosis. Cell Death Dis 17, 246 (2026). https://doi.org/10.1038/s41419-026-08493-8
Słowa kluczowe: stłuszczenie wątroby, mitochondria, śmierć komórkowa, metabolizm żelaza, zapalenie wątroby