Clear Sky Science · pl
Celowanie w mitochondrialną fosfatazę PGAM5 łagodzi ferropto i ostre zapalenie trzustki poprzez zwiększenie ekspresji FSP1 zależnej od NRF2
Dlaczego zmęczone komórki i obolałe trzustki są ważne
Kiedy komórki są przeciążone, mogą ginąć w sposób, który szkodzi całemu organizmowi. Jedna z takich form śmierci komórkowej, zwana ferroptozą, napędzana jest przez żelazo i wymykające się spod kontroli reakcje chemiczne, które „zardzewiają” tłuszcze w błonach komórkowych. Proces ten wiąże się z uszkodzeniami narządów, w tym z bolesnym i niekiedy śmiertelnym stanem znanym jako ostre zapalenie trzustki. Badanie opisane w tym artykule ujawnia kluczowy przełącznik wewnątrz mitochondriów — elektrowni komórki — który może wzmacniać lub osłabiać ferroptozę, i pokazuje, że zablokowanie tego przełącznika może chronić trzustkę w modelu mysim.
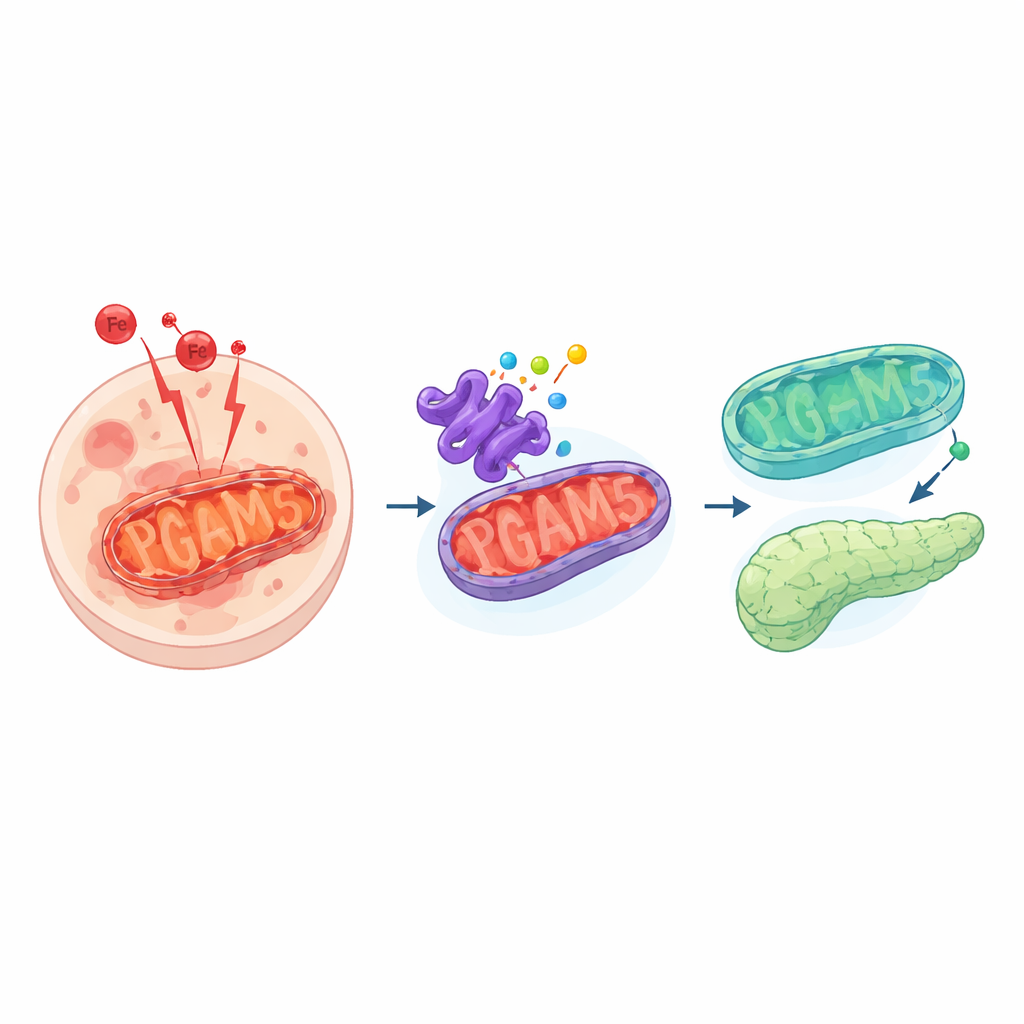
Niebezpieczna forma śmierci komórkowej
Ferroptoza różni się od bardziej znanych form śmierci komórkowej, takich jak apoptoza. Zamiast uporządkowanej autodestrukcji, komórki przechodzące ferroptozę doświadczają burzy reakcji napędzanych przez żelazo, które utleniają wrażliwe lipidy w ich błonach. Powstają toksyczne produkty uboczne i ubytki w błonach, które ostatecznie prowadzą do śmierci komórki. Zazwyczaj komórki kontrolują to mechanizmami ochronnymi, które detoksykują reaktywne molekuły. Gdy te systemy zawodzą lub są przytłoczone, ferroptoza może rozsiewać uszkodzenia w tkankach, przyczyniając się do chorób od raka po niewydolność narządów.
Mitochondrialny przełącznik w oku cyklonu
Naukowcy skupili się na białku PGAM5, które znajduje się po wewnętrznej stronie mitochondriów i działa jako centrum sygnalizacyjne. PGAM5 pomaga kontrolować kształt mitochondriów, reaguje na stres i wpływa na to, jak komórki radzą sobie z utlenianiem. Co zaskakujące, gdy zespół albo zmniejszał poziom PGAM5, albo wymuszał jego nadprodukcję, komórki stawały się trudniejsze do zabicia przez ferroptozę. Chemiczna inhibicja PGAM5, genetyczne stłumienie oraz nadekspresja zmniejszały nagromadzenie szkodliwych produktów utleniania lipidów i ograniczały śmierć komórek wywołaną przez lek indukujący ferroptozę. To ujawniło, że system jest precyzyjnie wyregulowany: zarówno zbyt mało, jak i zbyt dużo PGAM5 przesuwa komórki w kierunku bardziej chronionego stanu.
Włączenie wewnętrznej tarczy
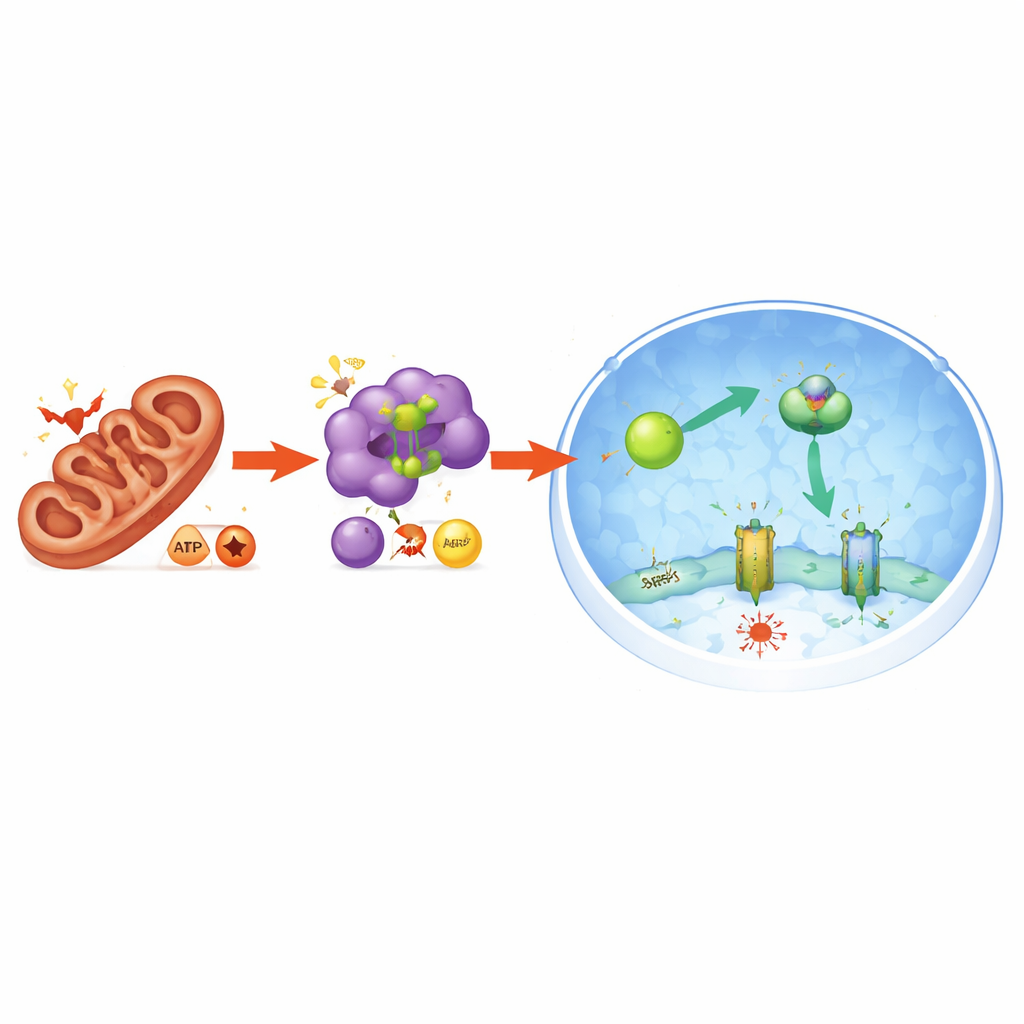
Pogłębiając analizę, autorzy odkryli, że wpływ PGAM5 przebiega przez ochronną oś obejmującą dwóch innych uczestników: NRF2 i FSP1. NRF2 to główny regulator, który po aktywacji i wejściu do jądra włącza szeroki zestaw mechanizmów antyoksydacyjnych. FSP1 jest jednym z jego efektorów, pomagającym regenerować rozpuszczalny w tłuszczach antyoksydant, co blokuje uszkodzenia lipidów w błonie komórkowej. Gdy poziomy PGAM5 były modyfikowane, komórki zwiększały zarówno poziom mRNA, jak i białka NRF2, a NRF2 łatwiej przemieszczał się z cytoplazmy do jądra. Tam nasilał produkcję FSP1. Zablokowanie FSP1 lub NRF2 likwidowało ochronę i przywracało wrażliwość na ferroptozę, co dowodzi, że łańcuch PGAM5–NRF2–FSP1 jest niezbędny dla zaobserwowanej odporności.
Stres energetyczny jako ukryty przekaźnik
Badanie ujawniło również, jak stres mitochondrialny przekłada się na tę ochronną reakcję. Zakłócenie PGAM5 zaburzało równowagę mitochondriów i zmieniało „walutę energetyczną” komórki: wzrósł stosunek niskoenergetycznych nukleotydów (AMP i ADP) do wysokoenergetycznego ATP, co sygnalizowało stres energetyczny. To z kolei aktywowało enzym wyczuwający energię — AMPK. Aktywowany AMPK bezpośrednio modyfikował NRF2 w sposób sprzyjający jego wejściu do jądra, co dodatkowo zwiększało produkcję FSP1. Usunięcie AMPK z systemu uniemożliwiało nagromadzenie NRF2 w jądrze, spadały poziomy FSP1 i komórki ponownie ulegały ferroptozie. W ten sposób PGAM5 łączy stan mitochondriów z szerszą odpowiedzią energetyczną i antyoksydacyjną, która chroni komórki przed żelazem napędzaną śmiercią.
Ochrona trzustki in vivo
Aby sprawdzić, czy ten mechanizm ma znaczenie dla całego narządu, badacze użyli modelu mysiego ostrego zapalenia trzustki wywołanego wysokimi dawkami aminokwasu argininy. W tym modelu trzustka wykazywała rozległe uszkodzenia, podwyższone poziomy enzymów we krwi świadczące o urazie tkanki oraz wzrost stężenia cząsteczek zapalnych. Markery peroksydacji lipidów — sygnatura ferroptozy — również wyraźnie wzrosły w trzustce. Leczenie myszy związkiem hamującym PGAM5 łagodziło te objawy: spadły markery uszkodzenia we krwi, tkanka trzustkowa wyglądała w mikroskopie zdrowiej, a sygnały zapalne zmalały. Równocześnie zmniejszyły się markery ferroptozy, a w trzustce wzrosła aktywność AMPK oraz poziomy NRF2 i FSP1, co odpowiadało ochronnej ścieżce zaobserwowanej w hodowlach komórkowych.
Co to znaczy dla przyszłych terapii
Podsumowując, praca wskazuje PGAM5 jako centralny punkt kontrolny łączący stres mitochondriów, status energetyczny komórki i silny program antyoksydacyjny blokujący ferroptozę. Poprzez osłabienie aktywności PGAM5 komórki aktywują AMPK i NRF2, zwiększają FSP1 i lepiej znoszą uszkodzenia lipidów napędzane żelazem. U myszy ta strategia zmniejsza uraz trzustki w ostrym zapaleniu. Dla czytelnika popularnonaukowego przekaz jest taki: naukowcy odkryli nowy wewnętrzny „wyłącznik obwodu”, który może zapobiegać destrukcyjnej formie śmierci komórkowej. Choć przed zastosowaniem klinicznym pozostało wiele pracy, celowanie w PGAM5 lub jego późniejsze ogniwa może otworzyć nowe drogi leczenia schorzeń, w których ferroptoza i niewydolność mitochondrialna odgrywają szkodliwą rolę.
Cytowanie: Ma, S., Qin, J., Luan, J. et al. Targeting mitochondrial phosphatase PGAM5 alleviates ferroptosis and acute pancreatitis by upregulating NRF2-mediated FSP1 expression. Cell Death Dis 17, 252 (2026). https://doi.org/10.1038/s41419-026-08484-9
Słowa kluczowe: ferroptoza, mitochondria, ostre zapalenie trzustki, stres oksydacyjny, śmierć komórkowa