Clear Sky Science · pl
Semaforyna 6D wyzwala przeciwnowotworowe odpowiedzi typu I interferonu, przekształcając mikrośrodowisko guza w raku jelita grubego
Dlaczego te badania mają znaczenie dla osób z rakiem jelita
Rak jelita grubego jest jedną z głównych przyczyn zgonów z powodu nowotworów na świecie, częściowo dlatego, że wiele guzów opiera się wobec dostępnych terapii, w tym nowoczesnych leków immunoterapeutycznych. W tym badaniu odkryto naturalny „hamulec” wewnątrz nowotworów jelita, który często jest wyłączony, oraz pokazano, że jego ponowne włączenie może przyciągnąć układ odpornościowy z powrotem do walki. Zrozumienie tego ukrytego przełącznika kontrolnego może pomóc lekarzom lepiej przewidywać rokowanie i projektować terapie skojarzone, które uczynią immunoterapię skuteczną dla znacznie większej liczby pacjentów.
Cichy strażnik wewnątrz komórek nowotworowych
W centrum badań znajduje się cząsteczka nazwana Semaforyną 6D (SEMA6D), pierwotnie znana z prowadzenia wzrostu nerwów i kształtowania rozwijającego się serca. Badacze odkryli, że SEMA6D zachowuje się jak supresor nowotworowy w raku jelita grubego: w zdrowej tkance jelita jest obecna, lecz w tkance nowotworowej jej poziomy są wyraźnie obniżone. W wielu zestawach danych pacjentów i próbkach guzów niski poziom SEMA6D wiązał się z większymi guzami, głębszą inwazją, większą liczbą przerzutów oraz istotnie gorszym przeżyciem. Wzorzec ten utrzymywał się nawet po uwzględnieniu innych czynników klinicznych, co wskazuje, że SEMA6D jest niezależnym wskaźnikiem agresywności guza jelita grubego.
Dlaczego guzy wyłączają tę ochronę
Zespół następnie zapytał, dlaczego SEMA6D jest tak często nieobecna w guzach. Odkryto, że gen ten jest często wyciszany przez chemiczną modyfikację zwaną hypermetylacją promotora — dodatkowe znaczniki chemiczne dodane do regionu kontrolnego genu, działające jak molekularna taśma zakrywająca wyłącznik światła. Dzięki szczegółowemu mapowaniu DNA wykazano, że kluczowe odcinki regionu kontrolnego SEMA6D są silnie metylowane w komórkach nowotworowych, lecz nie w normalnych komórkach jelita. Gdy leczono komórki nowotworowe lekiem demetylującym stosowanym w nowotworach krwi, znaczniki metylowe zostały usunięte, a produkcja SEMA6D została przywrócona. Najniższe poziomy SEMA6D obserwowano w podtypach raka jelita już wcześniej powiązanych z intensywną metylacją DNA, dużą niestabilnością genetyczną i skłonnością do rozsiewu, co wzmacnia związek między tym mechanizmem wyciszania a agresywną chorobą.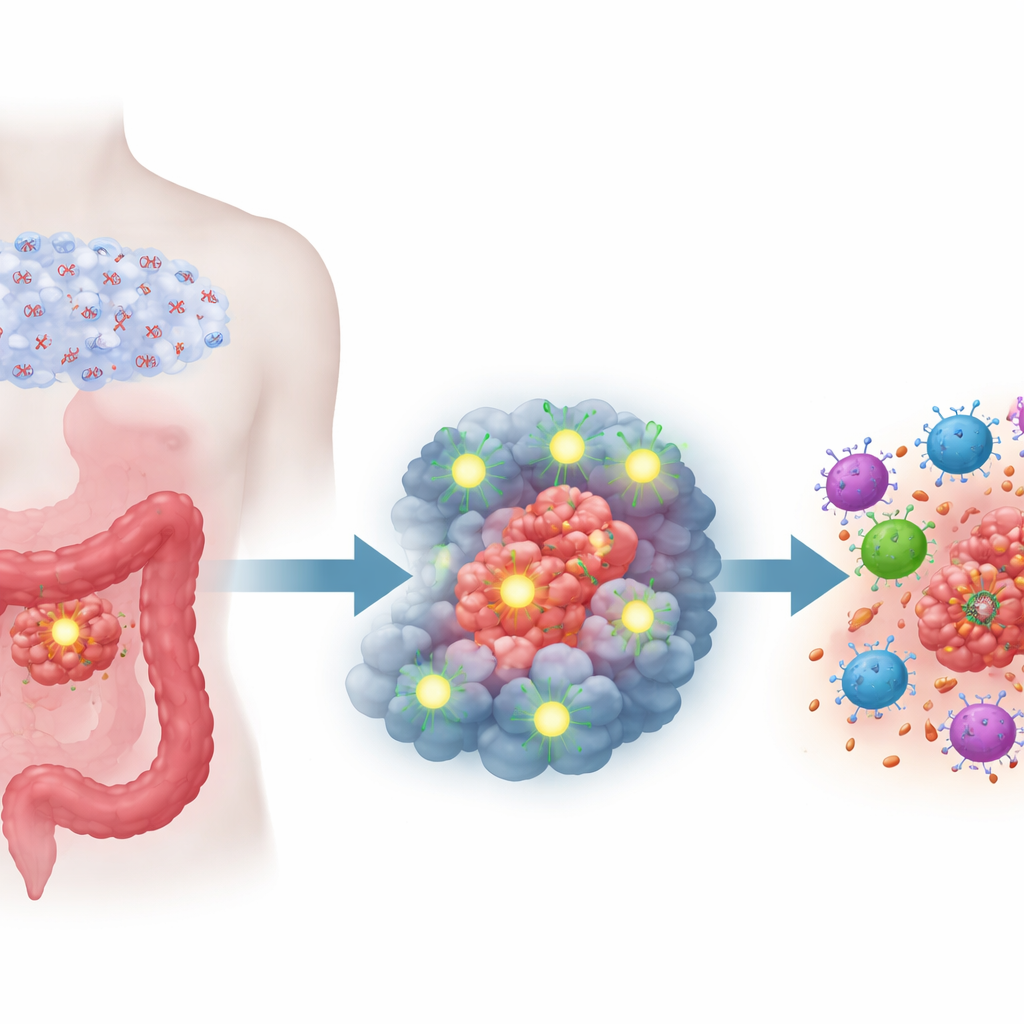
Od blokera wzrostu do wzmacniacza odporności
Przywrócenie SEMA6D zmieniło zachowanie guza na dwóch poziomach. Po pierwsze, na poziomie komórki nowotworowej — wymuszenie większej produkcji SEMA6D spowolniło ich wzrost, zmniejszyło zdolność migracji i inwazji oraz odwróciło cechy przejścia nabłonkowo-mezenchymalnego, programu sprzyjającego rozsiewowi guzów. W hodowlach i trójwymiarowych organoidach pochodzących z guzów pacjentów komórki z wyższą SEMA6D tworzyły mniej i mniejsze kolonie oraz wykazywały więcej oznak zaprogramowanej śmierci komórkowej. U myszy guzy zaprojektowane do nadprodukcji SEMA6D rosły wolniej i dawały mniej przerzutów do płuc i wątroby, podczas gdy obniżenie SEMA6D przynosiło efekt odwrotny. Po drugie, na poziomie odpornościowym — guzy bogate w SEMA6D u immunokompetentnych myszy zawierały znacznie więcej limfocytów T CD4 i CD8 — głównych sił ataku odporności adaptacyjnej — podczas gdy guzy ubogie w SEMA6D były stosunkowo pozbawione tych obrońców. Po usunięciu limfocytów T efekt spowalniający wzrost związany z SEMA6D w dużej mierze zanikał, co pokazuje, że wiele jej działania wynika z mobilizacji układu odpornościowego.
Odkodowanie wewnętrznej ścieżki alarmowej
Pogłębiając badania, zespół odwzorował kroki molekularne łączące SEMA6D z aktywacją odporności. Na powierzchni komórek nowotworowych SEMA6D sygnalizuje poprzez partnera-receptor zwanego Plexin A4. Wewnątrz komórki duet ten fizycznie współdziała z białkiem IRF9, kluczowym elementem maszynerii reagującej na interferony typu I — tych samych sygnałów alarmowych antywirusowych, które komórki wykorzystują do walki z infekcjami. Gdy SEMA6D jest obecna i Plexin A4 jest nienaruszony, IRF9 i jego partnerzy ulegają aktywacji, włączają zestawy genów stymulowanych interferonem i pomagają komórce nowotworowej nadawać sygnały, które przyciągają i uzbrajają limfocyty T. Usunięcie SEMA6D lub Plexin A4 przerywa ten łańcuch i przytłumia alarm; przywrócenie IRF9 może częściowo przywrócić efekt. U myszy guzy z aktywnym sygnalizowaniem SEMA6D–Plexin A4–IRF9 miały więcej infiltrujących limfocytów T i niższe poziomy markera proliferacji Ki-67, co jest zgodne ze zwiększoną presją immunologiczną na nowotwór.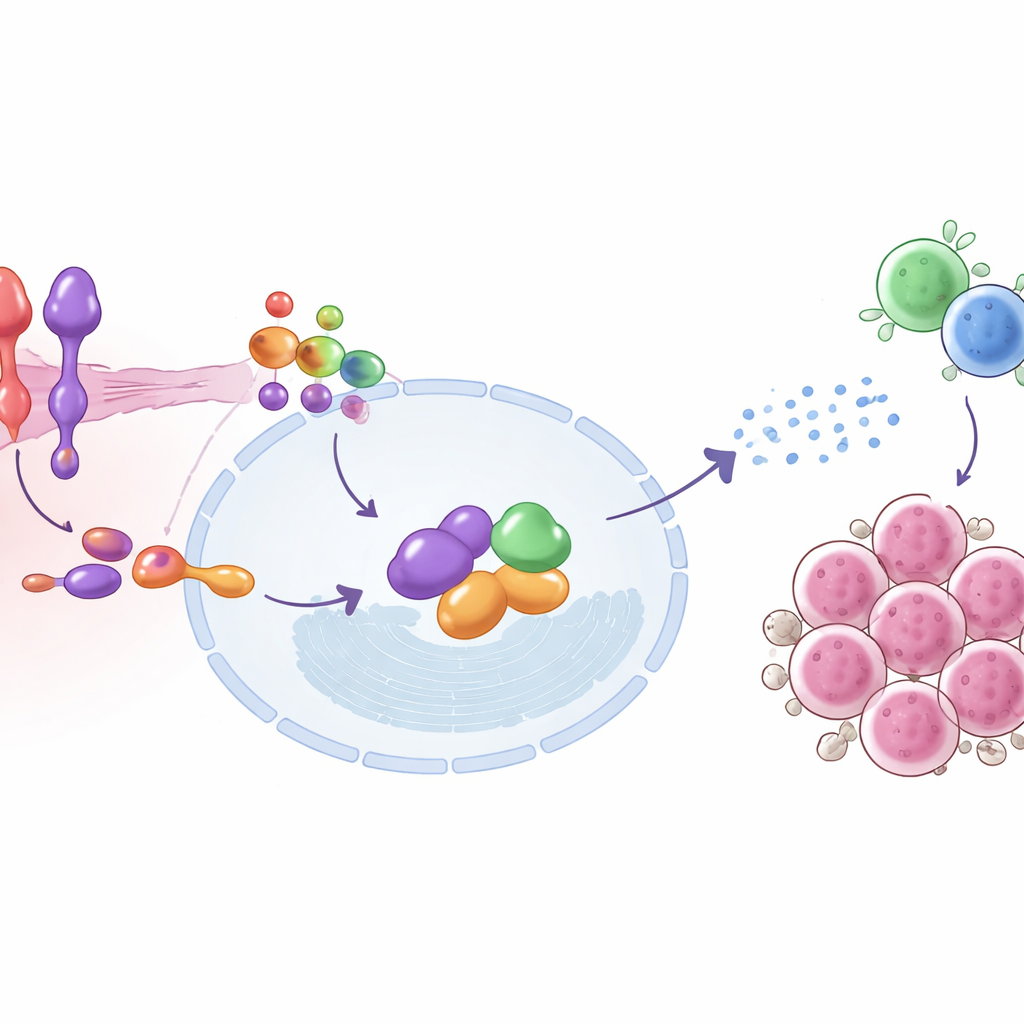
Przebudzenie odporności terapią skojarzoną
Ponieważ SEMA6D jest wyciszana przez metylację, autorzy sprawdzili, czy lek hypometylujący może reaktywować go w żywych guzach i poprawić odpowiedź na blokadę punktów kontrolnych odporności. W mysich guzach jelita leczonych dechemidyną (decitabine) połączoną z przeciwciałem anty–PD-1 guzy rosły znacznie wolniej niż po każdym z tych zabiegów osobno. Kombinacja zwiększyła poziomy SEMA6D, wzmocniła aktywność szlaku interferonowego, zmniejszyła proliferację komórek i zwiększyła infiltrację limfocytów T. Wyniki te sugerują, że przez usunięcie „zamków” metylacyjnych z genów istotnych dla odporności, takich jak SEMA6D, leki epigenetyczne mogą przekształcić immunologicznie „zimne” guzy w „cieplejsze”, bardziej podatne na inhibitory punktów kontrolnych.
Co to oznacza dla przyszłej opieki
Dla osoby niebędącej specjalistą wniosek jest taki, że niektóre raki jelita chowają się przed układem odpornościowym, chemicznie wyłączając wbudowany sygnał niebezpieczeństwa. Praca ta identyfikuje SEMA6D zarówno jako ten sygnał, jak i obiecujący cel terapeutyczny. Pomiar poziomu SEMA6D i statusu jej metylacji mógłby pomóc w klasyfikacji guzów, prognozowaniu wyników i kierowaniu wyborami terapeutycznymi. Równie ważne, badanie dostarcza jasnego biologicznego uzasadnienia dla łączenia środków demetylujących DNA z immunoterapią, aby przebudzić nadzór immunologiczny u pacjentów, których guzy obecnie nie reagują. Choć nadal potrzebne są badania kliniczne, ta strategia może pewnego dnia rozszerzyć korzyści immunoterapii na znacznie większą grupę osób z rakiem jelita grubego.
Cytowanie: Shi, W., Zhang, F., Sun, WQ. et al. Semaphorin 6D drives anti-tumor type I interferon responses to reprogram the tumor microenvironment in colorectal cancer. Cell Death Dis 17, 256 (2026). https://doi.org/10.1038/s41419-026-08478-7
Słowa kluczowe: rak jelita grubego, mikrośrodowisko guza, terapia epigenetyczna, interferon typu I, immunologia nowotworów