Clear Sky Science · pl
Warianty powodujące utratę funkcji w HPDL upośledzają rozwój kory mózgowej u człowieka poprzez zaburzenia funkcji mitochondriów
Dlaczego małe „silniki” komórkowe mają znaczenie dla rozwijającego się mózgu
Większość osób, myśląc o rozwoju mózgu, wyobraża sobie genetykę i połączenia nerwowe. To badanie pokazuje, że inny, często pomijany czynnik — niewielkie elektrownie w naszych komórkach, czyli mitochondria — również mogą kształtować sposób, w jaki formuje się mózg. Analizując rzadkie dziecięce zaburzenia ruchowe związane z genem HPDL, autorzy ujawniają, jak zaburzenia produkcji energii mogą zmniejszać rozwijającą się korę mózgową, strukturę kluczową dla ruchu, myślenia i zachowania.
Rzadkie zaburzenie ruchowe jako okno na wzrost mózgu
U niektórych dzieci ze zmianami w genie HPDL rozwija się dziedziczna spastyczna paraplegia, stan powodujący sztywność i osłabienie nóg, a także napady padaczkowe, opóźnienie rozwoju i w cięższych przypadkach mniejszy niż normalnie mózg (mikrocefalia). Chociaż wiadomo było, że białko HPDL lokalizuje się w mitochondriach, jego dokładna rola — i dlaczego jego utrata uszkadza mózg — była niejasna. Badacze użyli kilku modeli komórek ludzkich, w tym komórek nowotworowych o cechach nerwowych oraz komórek mózgowych wyhodowanych z próbek skóry pacjentów, aby sprawdzić, czy HPDL jest potrzebny do prawidłowego rozwoju mózgu i funkcji mitochondriów.
Co się dzieje, gdy HPDL jest wyłączony
Początkowo zespół wyłączył HPDL w ludzkiej linii komórkowej neuroblastomy za pomocą edycji genów CRISPR. Bez HPDL komórki straciły pełnej długości białko i wykazywały wyraźne problemy mitochondrialne. Duże zespoły białek łańcucha oddechowego, które normalnie współpracują przy wytwarzaniu energii, zostały zaburzone, a kluczowe składniki związane z wykorzystaniem tlenu były zmniejszone. Komórki używały mniej tlenu, generowały mniej oddechu powiązanego z produkcją energii i wytwarzały więcej reaktywnych form tlenu — szkodliwych produktów ubocznych zwanych „stresem oksydacyjnym”. Jednak całkowita liczba mitochondriów nie zmalała, a poziomy koenzymu Q10, ważnej cząsteczki dla transferu energii, były wręcz wyższe, co sugeruje defekt jakościowy — nie tylko ilościowy — funkcji mitochondriów.
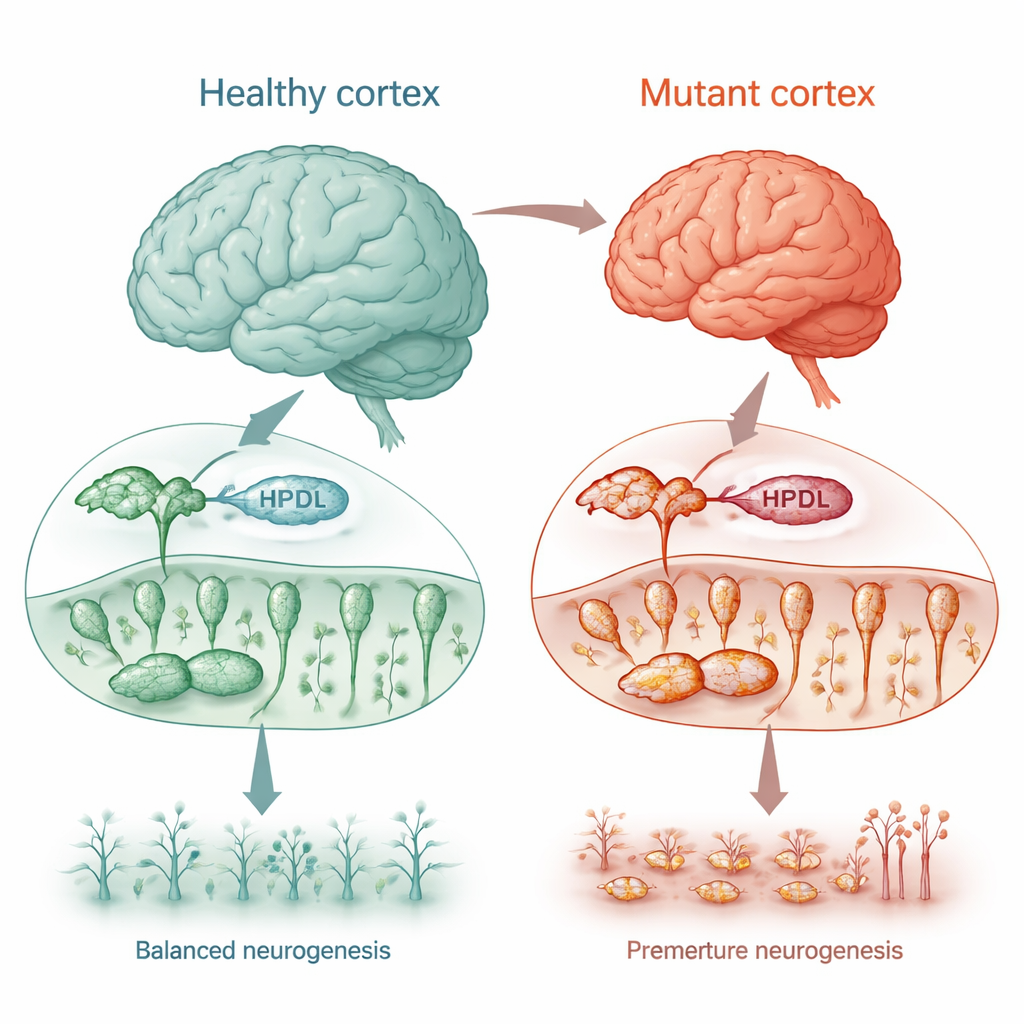
Tkanka mózgowa w probówce ujawnia wczesne nadprodukcje neuronów
Aby sprawdzić, jak utrata HPDL wpływa na prawdziwy rozwój ludzkiego mózgu, badacze przekształcili komórki skóry od czterech dotkniętych dzieci w indukowane komórki pluripotentne, a następnie skłonili je do formowania komórek korowych i trójwymiarowych „mini-mózgów” (organoidów). We wczesnym stadium rozwoju, w momencie gdy większość komórek powinna jeszcze dzielić się jako progenitory nerwowe, hodowle z mutantem HPDL już zawierały więcej dojrzałych neuronów i mniej progenitorów. Profile aktywności genów potwierdziły to: szlaki napędzające powstawanie neuronów zostały włączone zbyt wcześnie, podczas gdy te utrzymujące komórki w stanie proliferacji były przyciszone. W organoidach ta przedwczesna zmiana od „cegiełek budulcowych” do dojrzałych neuronów doprowadziła do znacznie mniejszych struktur przypominających mózg, co odzwierciedla mikrocefalię obserwowaną u najciężej dotkniętych dzieci.
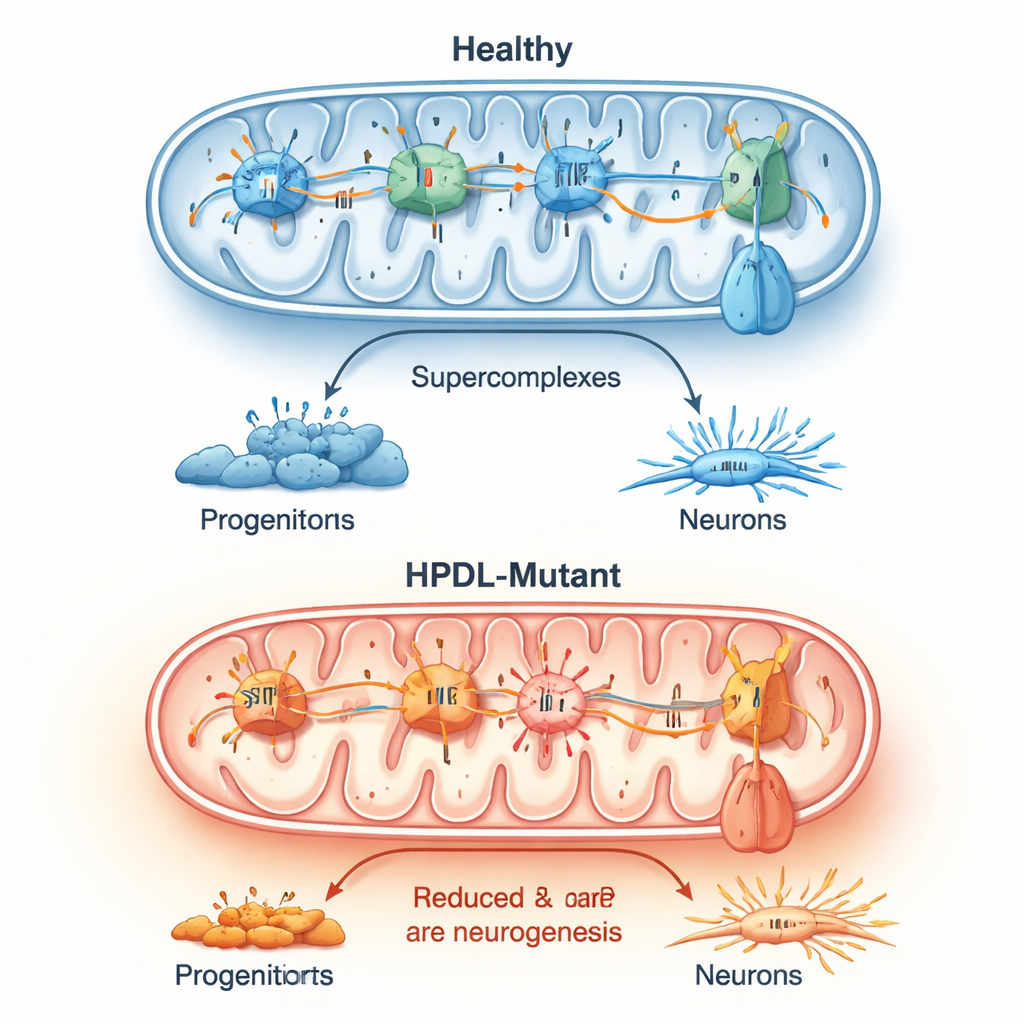
Uszkodzone elektrownie i zestresowane komórki
Bardziej szczegółowe badania wykazały, że komórki mózgowe z mutantem HPDL miały upośledzoną fosforylację oksydacyjną — główny sposób, w jaki mitochondria wytwarzają energię. Barwienia enzymatyczne ujawniły słabszą aktywność kluczowego kompleksu mitochondrialnego, a inne miary wykazały zmienione napięcie elektryczne na błonie mitochondrialnej. W wielu komórkach mutantów istotny enzym zwykle produkujący ATP wydawał się pracować „do tyłu”, by podtrzymać to napięcie błonowe — oznaka głębokiego zaburzenia metabolicznego. We wszystkich liniach pacjentów poziomy reaktywnych form tlenu były konsekwentnie podwyższone, a normalne duże zespoły białek łańcucha oddechowego były słabiej uformowane. Te zmiany mitochondrialne ściśle korelowały z czasem i stopniem przedwczesnej produkcji neuronów.
Testowanie sposobów łagodzenia stresu
Ponieważ stres oksydacyjny i zaburzona chemia koenzymu Q10 wydawały się kluczowe, zespół sprawdził, czy terapie ukierunkowane na te problemy mogą spowolnić pośpiech w kierunku tworzenia się neuronów. Narażono wczesne hodowle korowe na dwa przeciwutleniacze oraz na 4-hydroksybenzoesan, małą cząsteczkę związaną z syntezą koenzymu Q10. W kilku liniach pochodzących od pacjentów związki te częściowo zmniejszyły przedwczesną neurogenezę, ale odpowiedź zależała od konkretnej mutacji w HPDL. Niektóre linie odpowiadały głównie na przeciwutleniacze, inne na prekursor koenzymu Q10, a jedna w ogóle nie reagowała. Ten zależny od mutacji wzorzec sugeruje, że w zaburzeniach związanych z HPDL mogą być potrzebne spersonalizowane strategie terapeutyczne.
Co to oznacza dla dzieci i przyszłych terapii
Mówiąc prosto, badanie pokazuje, że HPDL działa jak strażnik „cegiełek budulcowych” mózgu w czasie wczesnego rozwoju. Gdy HPDL zawodzi, mitochondria stają się niewydolne i nadmiernie zestresowane, co popycha komórki progenitorowe do przekształcania się w neurony zbyt wcześnie. Zasób komórek dzielących się się wyczerpuje, kora nie osiąga pełnego rozmiaru, a wzorce połączeń ulegają zmianie, przyczyniając się do problemów z ruchem i innych objawów. Częściowe przywrócenie za pomocą przeciwutleniaczy i związków związanych z koenzymem Q10 sugeruje, że dostosowanie równowagi energetycznej i stresu oksydacyjnego w komórkach mogłoby kiedyś pomóc dzieciom z mutacjami HPDL, a być może także innym pacjentom z mitochondrialnymi formami chorób mózgu.
Cytowanie: Baggiani, M., Desbats, M.A., Naef, V. et al. Loss of function variants in HPDL impair human cortical development via alterations of mitochondrial function. Cell Death Dis 17, 237 (2026). https://doi.org/10.1038/s41419-026-08476-9
Słowa kluczowe: HPDL, mitochondria, rozwój kory mózgowej, mikrocefalia, stres oksydacyjny