Clear Sky Science · pl
Dysfunkcja interneuronów GABAergicznych leży u podstaw zmienionych oscylacji sieciowych związanych z aktywnością epileptopodobną u myszy pozbawionych PPT1
Kiedy rytmy mózgu zawodzą
Napady nie są tylko nagłymi burzami aktywności mózgowej; często wynikają ze subtelnych zmian w sposobie komunikacji komórek nerwowych. W tym badaniu przyjrzywano się rzadkiej chorobie dziecięcej, chorobie CLN1, i postawiono proste pytanie o dalekosiężnych konsekwencjach: co dzieje się z wewnętrznymi „utrzymywaczami rytmu” mózgu, gdy brakuje jednego enzymu, zwanego PPT1? Śledząc te zmiany u myszy w czasie, badacze ukazują, jak małe, wczesne zaburzenia inhibicji mogą narastać i prowadzić do napadów oraz rozległych uszkodzeń mózgu.
Strażnicy równowagi mózgu
Nasz mózg opiera się na dwóch szerokich typach komórek nerwowych. Komórki pobudzające, takie jak neurony piramidalne w hipokampie, napędzają aktywność. Komórki hamujące, zwane interneuronami, działają jak hamulce, kontrolując tę aktywność i kształtując elektryczne rytmy mózgu. Wśród nich dwie ważne grupy to interneurony parwalbuminowe (PV+) i somatostatynowe (SST+). Pomagają one generować i koordynować rytmiczne fale mózgowe, takie jak oscylacje theta i gamma, które wspierają funkcje takie jak uczenie się i pamięć. W chorobie CLN1 dzieci tracą enzym PPT1, który normalnie usuwa grupy tłuszczowe z białek. Autorzy wykorzystali model myszy z tą samą mutacją, jaką mają pacjenci, aby zobaczyć, jak utrata wpływa na interneurony i rytmy mózgowe, które one współtworzą.
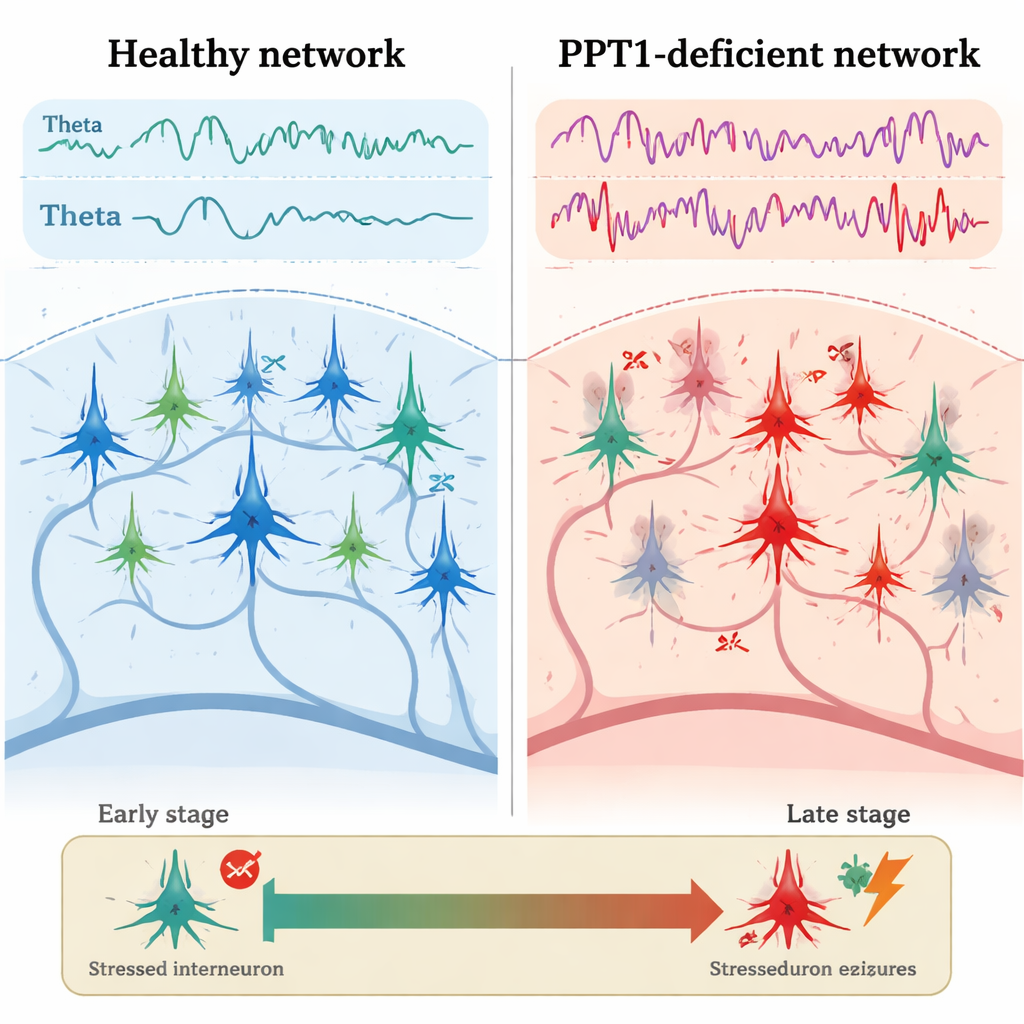
Wczesne pęknięcia w systemie hamowania
U młodych dorosłych myszy-mutantów, w wieku około trzech–czterech miesięcy, pierwszy wyraźny problem pojawił się w interneuronach PV+. Rejestracje elektryczne z hipokampa wykazały, że te komórki hamujące wyładowywały się rzadziej niż u zdrowych myszy, podczas gdy pobliskie neurony piramidalne wystrzeliwały częściej i z krótszymi przerwami między impulsami. Mikroskopia ujawniła, że wiele interneuronów PV+ aktywowało kaspazę-3, kluczowego wykonawcę programowanej śmierci komórkowej, mimo iż ich całkowita liczba jeszcze nie spadła. Równocześnie wzrosła moc fal theta i gamma, a obrazowanie wapniowe pokazało silniejszą aktywność neuronów hipokampa podczas poruszania się zwierząt. Co istotne, normalna „współzależność” między rytmami theta i gamma — gdzie wolniejsze fale pomagają organizować szybsze — osłabła, sugerując wczesny rozpad precyzyjnego timingu aktywności sieciowej.
Od zaburzonych rytmów do napadowych wyładowań
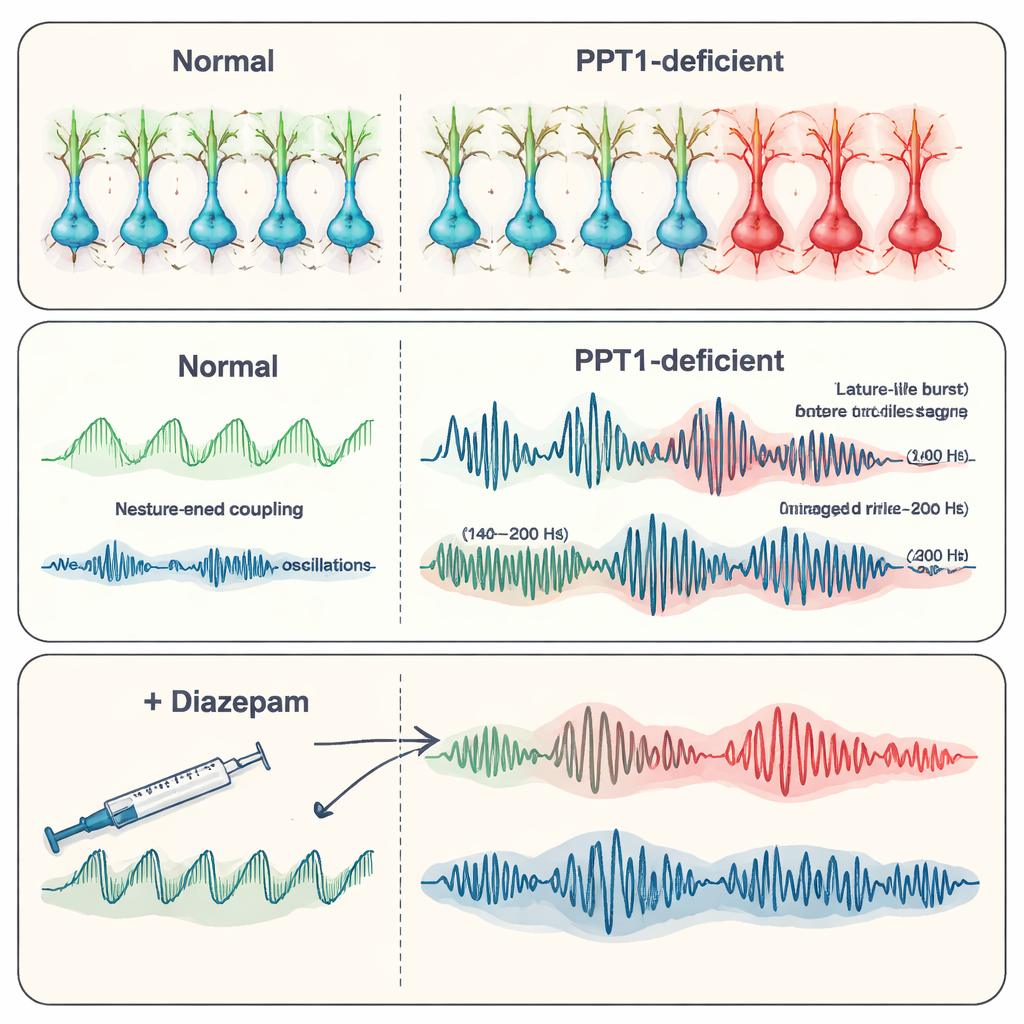
Do wieku sześciu–siedmiu miesięcy obraz pogorszył się. Wiele interneuronów PV+ uległo utracie, a teraz także interneurony SST+ wykazywały oznaki aktywacji kaspazy-3. Nagrania z hipokampa ujawniły spontaniczne wyładowania epileptopodobne — krótkie, nieprawidłowe wybuchy aktywności związane z napadami. Zespół skupił się na wysokoczęstotliwościowych „ripplach”, szybkich oscylacjach, które normalnie wspomagają konsolidację pamięci. U myszy-mutantów fizjologiczne ripplle (około 140–200 Hz) stały się rzadsze, ale o większej amplitudzie, podczas gdy jeszcze szybsze „patologiczne” ripplle (200–500 Hz), ściśle powiązane z padaczką, rosły na sile i częstotliwości. Razem te zmiany sugerowały przesunięcie od zorganizowanych rytmów związanych z pamięcią ku chaotycznym, podatnym na napady wzorom, gdy kontrola hamująca zawiodła.
Neurony zużywają się, a diazepam wkracza na scenę
W miarę postępu choroby sam hipokamp zaczął degenerować. Sygnały wapniowe w neuronach malały, barwienie Golgiego ujawniło cieńsze, mniej rozgałęzione drzewa dendrytyczne, a liczba małych kolców, gdzie tworzą się synapsy, zmniejszyła się. Zliczenia neuronów w kluczowych obszarach hipokampa (CA1 i CA3) potwierdziły rozległą utratę komórek, a w nagraniach elektrycznych rejestrowano mniej aktywnych jednostek. Badacze przetestowali następnie diazepam, powszechny lek przeciwdrgawkowy, który wzmacnia działanie hamującego neuroprzekaźnika GABA. U starszych myszy-mutantów diazepam zmniejszył częstość wyładowań epileptycznych i częściowo przywrócił bardziej normalne wzorce oscylacyjne, w tym zachowanie rippli, chociaż nie naprawił utraty receptorów. Sugeruje to, że wzmocnienie pozostałych sygnałów hamujących nadal może uspokoić sieć, przynajmniej tymczasowo.
Dlaczego te ustalenia są ważne
Dla czytelnika niebędącego specjalistą kluczowy przekaz jest taki, że choroba CLN1 to nie tylko gromadzenie odpadów w komórkach mózgowych. Utrata PPT1 uruchamia reakcję łańcuchową: najpierw wyspecjalizowane interneurony hamujące ulegają stresowi i zaczynają zawodzić, co uwalnia nadaktywne neurony piramidalne i zniekształca rytmy mózgowe. Z czasem ta nierównowaga prowadzi do napadów, a ostatecznie do masowej utraty komórek mózgowych i połączeń. Badanie wskazuje na okno możliwości we wczesnym stadium choroby, gdy ochrona lub ratowanie interneuronów PV+ — być może przez blokowanie aktywacji kaspazy — mogłoby zapobiec późniejszym napadom i degeneracji. Choć diazepam nie leczy CLN1, jego zdolność do tłumienia nieprawidłowych rytmów w tym modelu podkreśla szerszą ideę, że przywracanie hamowania może być silną strategią w leczeniu padaczki i związanych z nią zaburzeń mózgowych.
Cytowanie: Tong, J., Liu, W., Wang, Q. et al. Dysfunction of GABAergic interneurons underlies altered neural network oscillations associated with epileptiform activity in PPT1-deficient mice. Transl Psychiatry 16, 106 (2026). https://doi.org/10.1038/s41398-026-03843-8
Słowa kluczowe: padaczka, interneurony, hipokamp, oscylacje mózgowe, choroba spichrzania lizosomalnego