Clear Sky Science · pl
Zwiększanie aktywności KLF15 w kardiomiocytach: nowatorskie podejście zapobiegające patologicznej reprogramacji i włóknieniu za pomocą nukleazy-niewydajnego dCas9VPR
Reprogramowanie serca w niewydolności
Niewydolność serca dotyka miliony ludzi, rozwijając się często powoli po latach nadciśnienia lub choroby zastawek. W takich warunkach komórki mięśnia sercowego nie tylko powiększają się, lecz także włączają „płodowy” program genetyczny, a serce wypełnia się tkanką bliznowatą. W badaniu tym zbadano nowe sposoby subtelnego skierowania wewnętrznego mechanizmu kontroli genów serca z powrotem na tor zdrowia — bez cięcia DNA — poprzez delikatne zwiększenie aktywności ochronnego regulatora KLF15 w kardiomiocytach.
Gdy kardiomiocyty tracą tożsamość
W zdrowym dorosłym sercu kardiomiocyty — komórki mięśnia sercowego — efektywnie spalają lipidy na energię i utrzymują stabilny wzorzec aktywności genowej. Wykorzystując sekwencjonowanie RNA pojedynczych komórek u myszy poddanych przewlekłemu przeciążeniu ciśnieniem, badacze odwzorowali, jak poszczególne kardiomiocyty zmieniają się w miarę przechodzenia serca od prawidłowej funkcji do powiększenia i w konsekwencji do niewydolności. Stwierdzili, że czynnik transkrypcyjny KLF15, który normalnie utrzymuje równowagę między metabolizmem a wzrostem, wykazywał najsilniejszą zmianę aktywności w komórkach chorych. Wraz ze wzrostem stresu poziomy KLF15 spadały, a jego zdolność do tłumienia genów płodowych i związanych ze stresem osłabła. Podobne obniżenia KLF15 zaobserwowano w ludzkich sercach pacjentów z kardiomiopatią rozstrzeniową i przerostową, co wskazuje, że to zaburzenie jest zachowane między gatunkami.
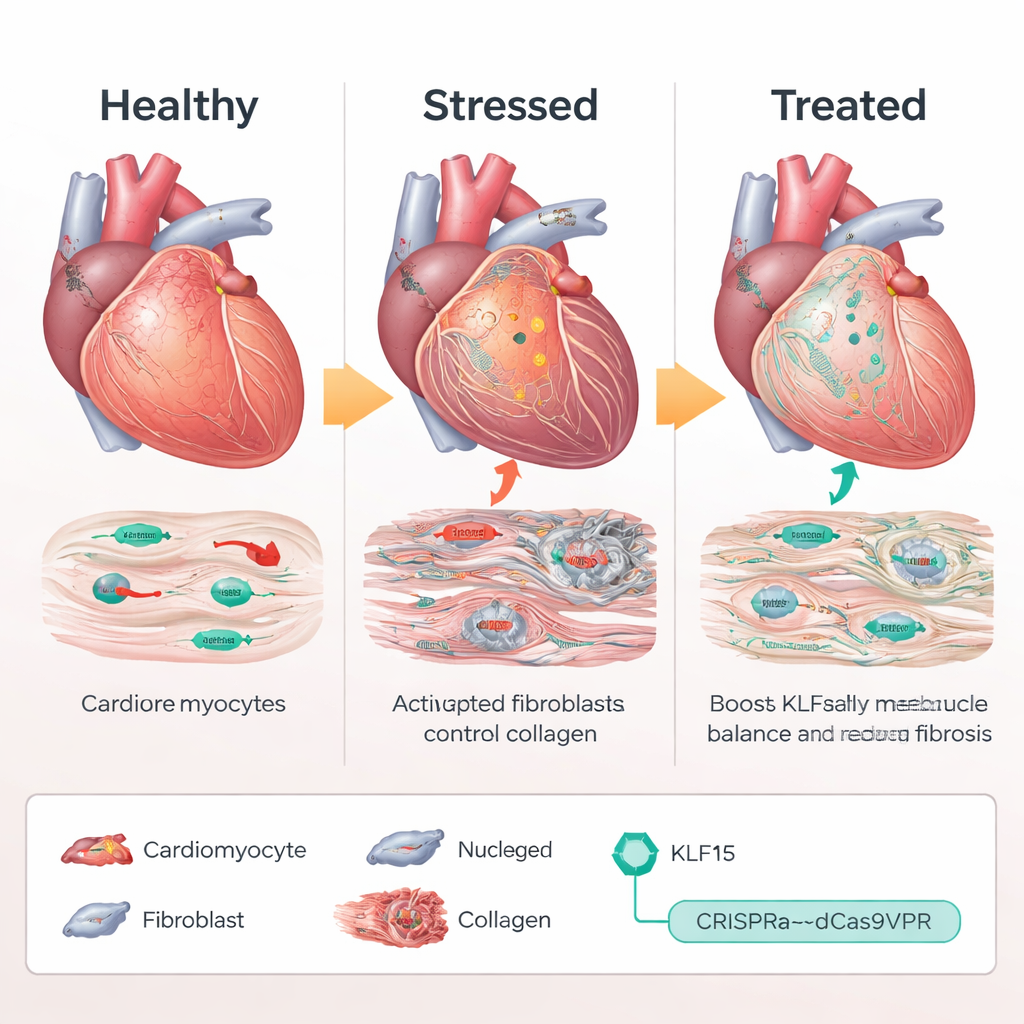
Wykorzystanie CRISPR jako pokrętła głośności, nie nożyc
Zamiast dodawać dodatkową kopię genu KLF15 lub przecinać DNA, zespół zastosował system „aktywacji” oparty na CRISPR, zwany dCas9VPR, który wiąże się w pobliżu naturalnego genu Klf15 i zwiększa jego ekspresję. U myszy zaprojektowanych tak, aby tę aktywatorkę CRISPR wyrażać tylko w kardiomiocytach, naukowcy dostarczyli RNA przewodniki za pomocą wirusa adeno-związanego (AAV9) skierowanego na promotor Klf15. Pod przewlekłym przeciążeniem ciśnieniowym myszy otrzymujące przewodniki aktywujące Klf15 utrzymywały niemal prawidłowe poziomy Klf15. Ich kardiomiocyty pozostawały mniejsze, funkcja wyrzutowa spadała w mniejszym stopniu, a przeżywalność poprawiła się w porównaniu z grupami kontrolnymi. Na poziomie molekularnym geny stresu i płodowe ucichły, podczas gdy geny metaboliczne i związane z obsługą wapnia odbudowały się, co wskazuje, że patologiczny program transkrypcyjny został w dużej mierze zresetowany.
Stłumienie tworzenia blizn przez komunikację międzykomórkową
Niewydolność serca jest napędzana nie tylko przez chore kardiomiocyty, ale także przez fibroblasty — komórki wspierające, które produkują kolagen i tworzą sztywną tkankę bliznowatą. Analizy pojedynczych komórek i obrazowanie tkanek wykazały, że przywrócenie Klf15 w kardiomiocytach zmniejszało aktywację fibroblastów i ogólne włóknienie, mimo że terapia genowa nigdy bezpośrednio nie celowała w fibroblasty. Zespół odnalazł źródło tego efektu w białku wydzielanym o nazwie AZGP1. Kiedy Klf15 było zwiększone w kardiomiocytach, produkcja i wydzielanie AZGP1 rosły. Zarówno w sercach myszy, jak i w tkankach sercowych pochodzących z ludzkich komórek macierzystych, wyższy poziom AZGP1 tłumił szlak TGF-β / SMAD w fibroblastach — kluczowy mechanizm napędzający bliznowacenie — obniżając poziomy markerów takich jak α-SMA i POSTN. Co istotne, nadekspresja AZGP1 jedynie w kardiomiocytach sama w sobie nie reprogramowała komórek mięśnia, co pokazuje, że KLF15 przede wszystkim chroni kardiomiocyty bezpośrednio i wykorzystuje AZGP1 jako posłańca do ograniczania aktywacji fibroblastów.
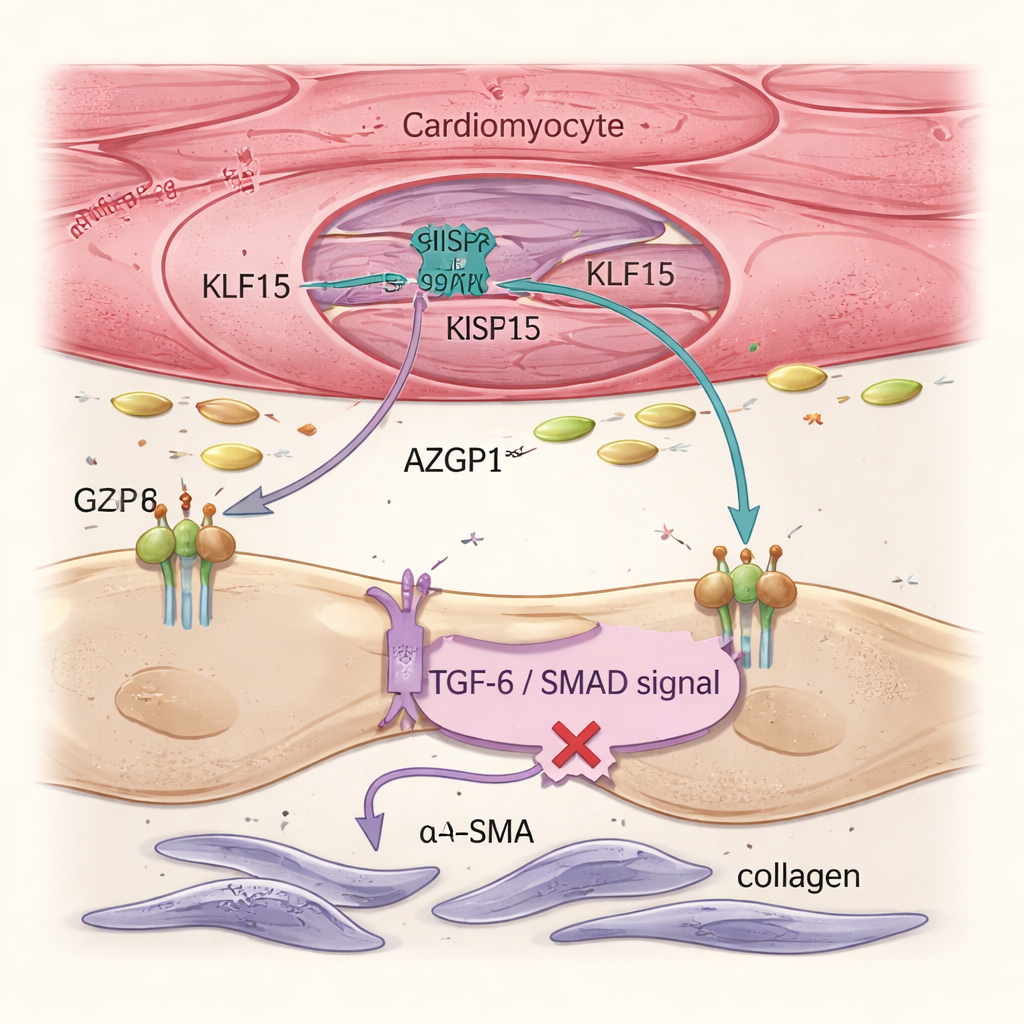
Modele tkankowe ludzkie potwierdzają ochronny obwód
Aby sprawdzić, czy te mechanizmy występują również w ludzkich komórkach, badacze użyli kardiomiocytów pochodzących z indukowanych pluripotentnych komórek macierzystych, hodowanych w trójwymiarowych inżynierowanych tkankach serca. Poddane obciążeniu mechanicznemu naśladującemu wysokie ciśnienie, tkanki te traciły KLF15, włączały geny stresu i płodowe, usztywniały się, a ich skurcze słabły — odtwarzając cechy choroby. Przywrócenie KLF15 za pomocą CRISPRa zapobiegło temu pogorszeniu, zachowało generowanie siły i przesunęło ekspresję genów z powrotem w kierunku dojrzałego metabolizmu i struktury. Szczegółowe eksperymenty wykazały, że TGF-β1, znany sygnał pro-włóknieniowy, obniża KLF15 w ludzkich kardiomiocytach poprzez szlak SMAD2/3, co pomaga wyjaśnić, jak przewlekły stres prowadzi do nieprzystosowawczej przebudowy. Wreszcie zespół zaprojektował kompaktowy, „mini” system CRISPRa oparty na mniejszym wariancie Cas9 mieszczącym się w pojedynczym wektorze AAV9 i napędzanym promotorem specyficznym dla kardiomiocytów. W precyzyjnie pociętych plasterkach niewydolnego ludzkiego mięśnia sercowego ten wektor skutecznie podnosił poziomy KLF15 i poprawiał wydolność kurczliwą w ciągu dni hodowli.
Plan delikatniejszej terapii genowej
Dla czytelnika niebędącego specjalistą główny przekaz jest taki, że praca ta pokazuje, jak ostrożne podkręcenie jednego ochronnego regulatora wewnątrz kardiomiocytów może zarówno ustabilizować ich tożsamość, jak i wysyłać sygnały ograniczające bliznowacenie. Dzięki zastosowaniu aktywatora CRISPR, który nie tnie DNA, podejście precyzyjnie dostraja naturalny gen serca zamiast wstawiać sztuczny. Badanie definiuje ścieżkę TGF-β → KLF15 → AZGP1 łączącą stres mechaniczny z szkodliwą przebudową i wykazuje, na modelach mysich, ludzkich komórkowych oraz plasterkach ludzkiej tkanki serca, że przywrócenie KLF15 może przerwać tę kaskadę. Choć nadal jest to etap przedkliniczny, kompaktowy, do kardiomiocytów ukierunkowany system CRISPRa przedstawiony tutaj oferuje potencjalną mapę drogową do leczenia powszechnych, niegenetycznych postaci niewydolności serca przez reprogramowanie aktywności genowej zamiast przepisywania genomu.
Cytowanie: Schoger, E., Kim, R., Bleckwedel, F. et al. Enhancing KLF15 activity in cardiomyocytes: a novel approach to prevent pathological reprogramming and fibrosis via nuclease-deficient dCas9VPR. Sig Transduct Target Ther 11, 76 (2026). https://doi.org/10.1038/s41392-026-02593-9
Słowa kluczowe: niewydolność serca, KLF15, aktywacja CRISPR, włóknienie serca, AZGP1