Clear Sky Science · pl
PMM2 wchodzi w interakcję z TRIM28, aby rekrutować E2F4 i promować KIFC3‑zależną glikolizę nowotworu oraz progresję raka jelita grubego
Dlaczego ta historia o raku ma znaczenie
Rak jelita grubego jest jednym z najbardziej śmiertelnych nowotworów na świecie, częściowo dlatego, że wiele guzów potrafi przejąć systemy energetyczne organizmu, by zasilać niekontrolowany wzrost. W tym badaniu odkryto, w jaki sposób mało poznany enzym PMM2 pomaga guzom jelita grubego intensywniej spalać cukier i się rozprzestrzeniać, oraz dlaczego czyni to z PMM2 obiecujący nowy cel dla przyszłych leków i testów diagnostycznych.
Silnik guza łaknący cukru
Komórki nowotworowe często przestawiają sposób wykorzystania glukozy, preferując szybkie, niskoefektywne spalanie cukru znane jako glikoliza. Badacze zaczęli od porównania tysięcy genów w próbkach nowotworów jelita grubego i w sąsiedniej zdrowej tkance. PMM2, enzym zwykle zaangażowany w dołączanie łańcuchów cukrowych do białek, wyróżnił się jako jeden z najsilniej podwyższonych genów w nowotworze. Komórki nowotworowe z nadmiarem PMM2 rosły szybciej, tworzyły więcej kolonii i łatwiej się rozprzestrzeniały w hodowlach, podczas gdy komórki z wyłączonym PMM2 spowalniały wzrost, migrowały mniej i częściej umierały.
Jak komórki nowotworowe turboładowują wykorzystanie cukru
Gdy zespół obniżył poziomy PMM2 w komórkach raka jelita grubego, komórki pobierały mniej glukozy, wytwarzały mniej ATP (głównej waluty energetycznej) i wydzielały mniej mleczanu, produktu ubocznego glikolizy. Wrażliwe pomiary metaboliczne potwierdziły, że ogólne zakwaszenie otaczającego podłoża spadło, natomiast zużycie tlenu wzrosło, co oznacza, że komórki przesunęły się z turboładowanej glikolizy w kierunku bardziej normalnego oddychania. Kluczowe białka wspomagające glikolizę, PKM2 i LDHA, także się obniżyły. Co zaskakujące, nawet katalitycznie „martwa” wersja PMM2 mogła nadal napędzać to łaknienie cukru, pokazując, że rola PMM2 w raku zależy nie od jego typowej aktywności enzymatycznej, lecz od tego, z kim się wiąże wewnątrz komórki.
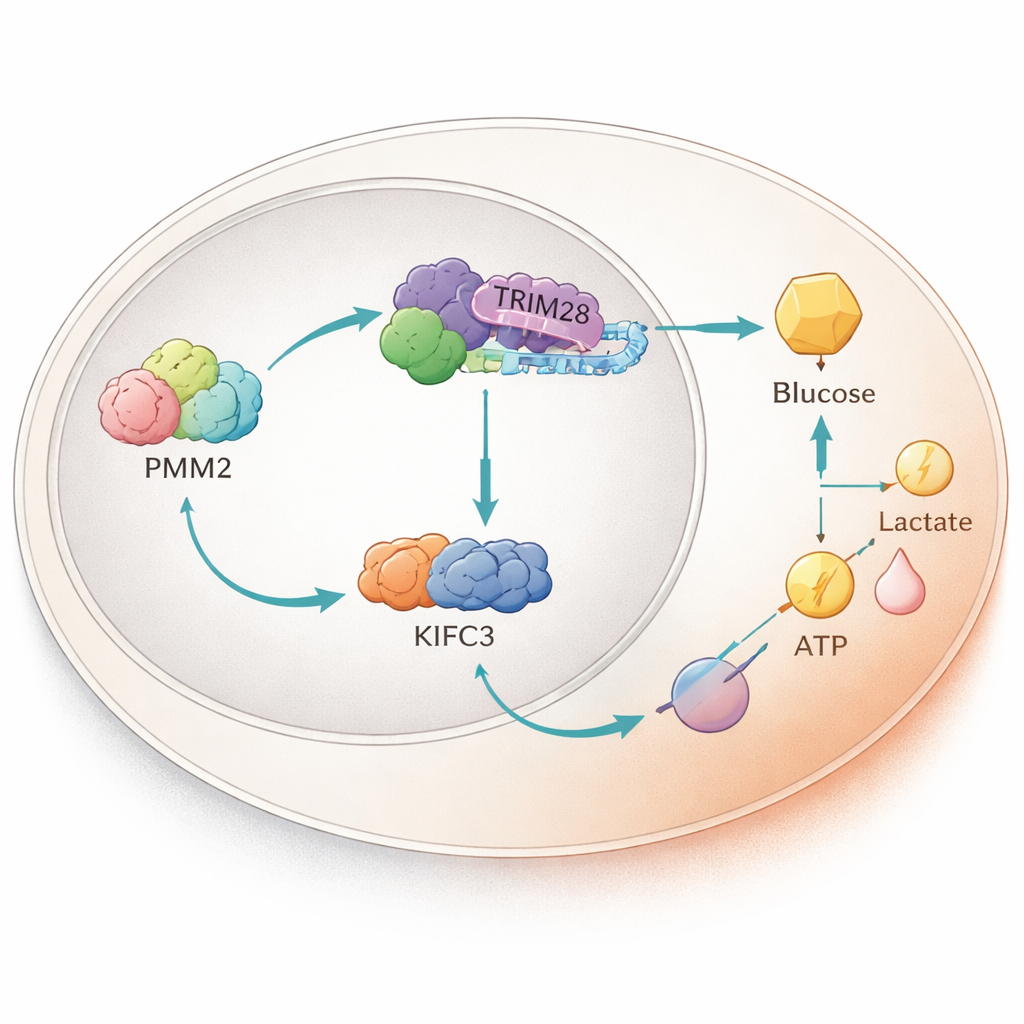
Przekaźnik białkowy wewnątrz jądra
Pogłębiając analizę, naukowcy odkryli, że PMM2 fizycznie łączy się z innym białkiem o nazwie TRIM28, które może przemieścić się do jądra komórkowego i wpływać na aktywność genów. PMM2 pomaga gromadzić TRIM28 w jądrze, gdzie TRIM28 współpracuje z czynnikiem transkrypcyjnym E2F4. Razem ta trójka zwiększa produkcję białka motorycznego KIFC3 przez wiązanie się z określonym fragmentem jego regionu kontrolnego DNA. Eksperymenty, które usunęły region PMM2 niezbędny do wiązania TRIM28, pozbawiły PMM2 zdolności do zwiększania glikolizy i wzrostu komórek, podkreślając, że to partnerstwo białkowe — a nie klasyczna funkcja enzymatyczna PMM2 — napędza przewagę guza.
Włączenie kluczowego przełącznika metabolicznego
KIFC3, znany głównie z roli w transporcie ładunków wzdłuż wewnętrznego rusztowania komórkowego, okazał się istotnym przełącznikiem metabolicznym. Gdy badacze obniżyli poziomy KIFC3, komórki raka jelita grubego zużywały mniej glukozy, produkowały mniej ATP i mleczanu oraz wykazywały słabszą aktywność glikolityczną, podczas gdy ich zużycie tlenu wzrosło. Co ważne, wygaszenie KIFC3 częściowo znosiło wzrost glikolizy i przewagę wzrostu wywołane przez PMM2. U myszy z implantowanymi ludzkimi komórkami raka jelita grubego guzy z nadmiarem PMM2 rosły większe, ale efekt ten był osłabiony, gdy KIFC3 został stłumiony. Próbki guzów od tych zwierząt wykazywały wyższe poziomy PMM2, KIFC3 i markerów glikolizy, łącząc cały łańcuch zdarzeń w tkance żywej.
Od modeli laboratoryjnych do próbek pacjentów
Aby przybliżyć badania do kliniki, zespół stworzył miniaturowe trójwymiarowe guzy, znane jako organoidy, z materiału pobranego od pacjentów z rakiem jelita grubego. Organoidy o wyższych poziomach PMM2 i KIFC3 rosły szybciej i wytwarzały więcej ATP oraz mleczanu niż te o niższych poziomach. Wymuszenie nadprodukcji PMM2 w organoidach zwiększało KIFC3 i glikolizę, podczas gdy redukcja PMM2 przynosiła odwrotne efekty. Analizy matryc nowotworowych od pacjentów wykazały ponadto, że wysokie poziomy PMM2 wiążą się z zaawansowaną chorobą, przerzutami do węzłów chłonnych i krótszym całkowitym przeżyciem, czyniąc z PMM2 silnego kandydata na biomarker.
Co to oznacza dla przyszłej opieki
W prostych słowach, to badanie pokazuje, że wiele guzów jelita grubego wydaje się włączać PMM2 do jądrowego przekaźnika białkowego — za pośrednictwem TRIM28 i E2F4 — aby zwiększyć KIFC3, a tym samym swój mechanizm spalania cukru. Ten metaboliczny przypływ pomaga nowotworom rosnąć i się rozprzestrzeniać. Ponieważ ta droga zależy od interakcji białkowych, a nie od zwykłej roli enzymatycznej PMM2, otwiera nowe możliwości terapeutyczne: małe cząsteczki, peptydy lub leki degradowane, które zaburzą wiązanie PMM2 z TRIM28, zablokują dostęp E2F4 do DNA lub stłumią aktywność KIFC3, mogłyby w zasadzie pozbawić guzy ich preferowanego paliwa. Chociaż takie terapie nie są jeszcze dostępne, łańcuch PMM2–TRIM28–E2F4–KIFC3 wyróżnia się teraz jako obiecująca mapa drogowa dla bardziej precyzyjnych strategii ukierunkowanych na metabolizm w walce z rakiem jelita grubego.
Cytowanie: Peng, Z., Ma, B., Song, Z. et al. PMM2 interacts with TRIM28 to recruit E2F4 and promote KIFC3-mediated tumor glycolysis and colorectal cancer progression. Oncogene 45, 1145–1160 (2026). https://doi.org/10.1038/s41388-026-03707-x
Słowa kluczowe: rak jelita grubego, metabolizm nowotworu, glikoliza, sygnalizacja onkogenna, marker