Clear Sky Science · nl
Energiezuinig wetenschappelijk rekenen met chemische reservoirs
Waarom chemie omzetten in rekenen ertoe doet
Moderne supercomputers verbruiken enorme hoeveelheden elektriciteit om het klimaat te simuleren, nieuwe geneesmiddelen te ontwerpen of kunstmatige intelligentie te trainen. Nu we de fysieke grenzen van traditionele chips naderen, wordt het steeds moeilijker en duurder om meer prestaties per watt te behalen. Dit artikel verkent een radicaal andere weg: echte chemische reacties gebruiken als de motor van wetenschappelijk rekenen. Door moleculen en hun interacties te behandelen als de bewegende onderdelen van een computer, schetsen de auteurs hoe toekomstige machines complexe vergelijkingen met veel minder energie kunnen oplossen dan de huidige digitale hardware.
Van levende cellen naar chemische rekenmachines
Levende cellen zijn meesterlijke probleemoplossers. Ze balanceren continu duizenden reacties om zich aan te passen, te groeien en te overleven, terwijl ze opmerkelijk weinig energie gebruiken. In het hart van dit gedrag bevinden zich chemische reactienetwerken—onderling verbonden reacties waarvan de snelheden en concentraties in de tijd veranderen. Deze netwerken kunnen worden beschreven met gewone differentiaalvergelijkingen, dezelfde wiskundige taal die gebruikt wordt om alles te modelleren, van epidemieën tot turbulente stromingen. De kerninzichten van dit werk zijn dat als chemie deze vergelijkingen al volgt, we die rechtstreeks zouden kunnen gebruiken om de berekeningen uit te voeren die wetenschappers nu op siliciumchips doen.
Hoe vergelijkingen reactienetwerken worden
De auteurs introduceren ChemComp, een softwareframework dat een stelsel differentiaalvergelijkingen systematisch omzet in een abstract netwerk van reacties. ChemComp gebruikt moderne compilatortechnologie om een wiskundig probleem op te delen in patronen die door geïdealiseerde reacties kunnen worden voorgesteld, en organiseert die vervolgens in een netwerk met duidelijk gedefinieerde soorten, verbindingen en snelheden. Deze abstracte reacties komen nog niet overeen met echte moleculen, maar vormen een blauwdruk voor een chemische computer. Het framework kan daarna biochemische reactiedatabases doorzoeken om echte reactiemotieven te vinden die zich vergelijkbaar gedragen, waarbij de voorkeur uitgaat naar opties die praktisch, veilig en mogelijk energiezuinig zijn in een laboratoriumomgeving.
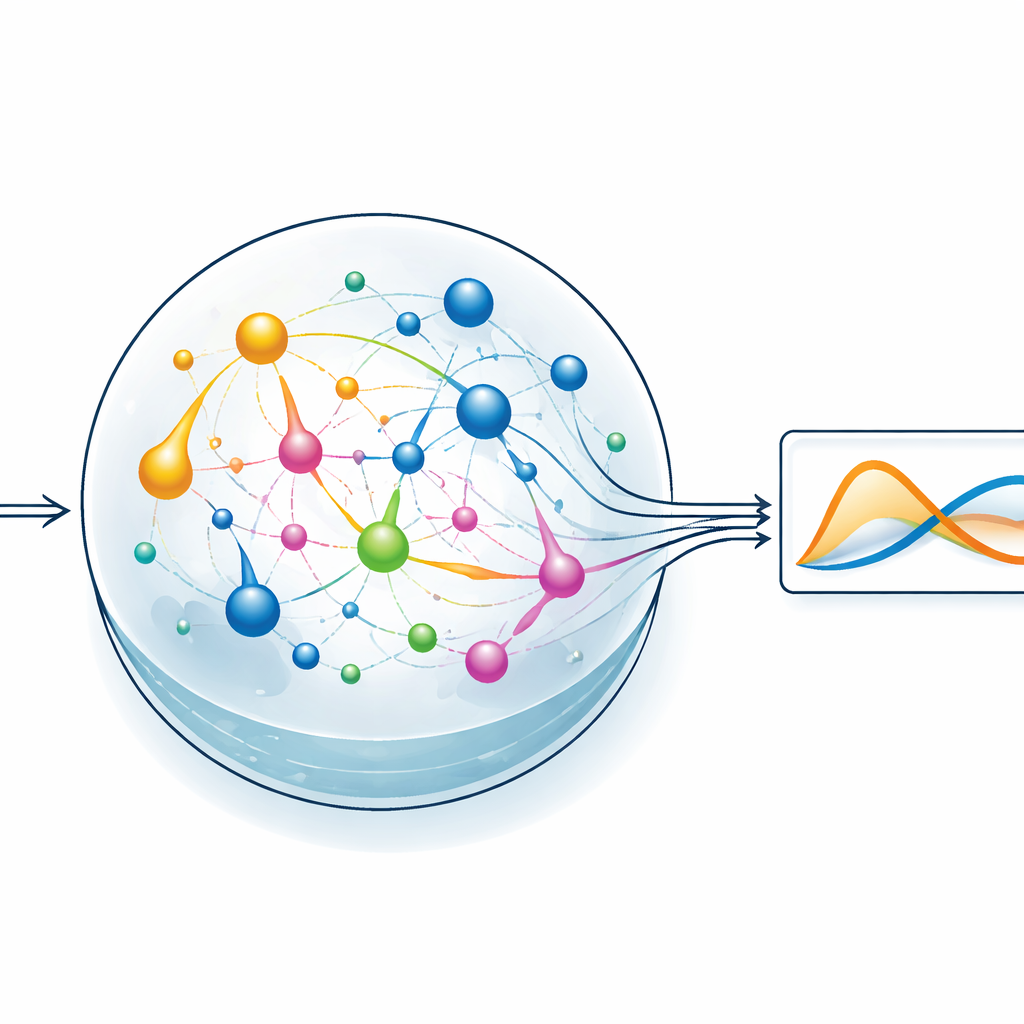
Een chemisch reservoir het zware werk laten doen
Om het idee te testen richt het team zich op een stijl van machine learning die reservoir computing heet. Hierbij transformeert een vast, dynamisch systeem een ingangssignaal naar een rijk, verward patroon van interne activiteit, en wordt alleen een eenvoudige uitleagelaag getraind om de gewenste uitvoer te produceren. In ChemComp’s uitvoering is het reservoir een set reacties in een goed geroerde vat; de veranderende concentraties van chemicaliën vormen de interne toestanden. De auteurs compileren een klassiek tweewaardig systeem bekend als het Sel’kov–Schnakenberg-model—oorspronkelijk gebruikt om oscillaties in de stofwisseling te bestuderen—tot kandidaat-reactienetwerken. Ze simuleren vervolgens hoe deze netwerken in de loop van de tijd reageren wanneer ze worden aangedreven door stromen van chemicaliën in en uit het vat, en gebruiken eenvoudige lineaire regressie om de concentratietraces te combineren tot een benadering van de doelfunctie.
Testen van eenvoudige en rijkere chemische netwerken
De onderzoekers vergelijken twee kandidaat-reservoirs: één met slechts twee chemische soorten en twee reacties, en een andere met vijf soorten en vijf reacties. Beide netwerken krijgen geschikte beginkoncentraties en stromingssnelheden, en worden vervolgens gesimuleerd terwijl ze draaien. Zelfs het kleinere systeem kan het oscillerende gedrag van de doelvergelijkingen grofweg reproduceren, maar het grotere netwerk doet het merkbaar beter en vermindert de fout zowel tijdens training als testen. Door te variëren in beginkoncentraties en reactiesnelheidsconstanten, brengen de auteurs gebieden in kaart waar het chemische systeem het meest nauwkeurig de gewenste dynamiek benadert. Elke reactie werkt in feite als een basisfunctie in een curve-fit-probleem: hoe gevarieerder de beschikbare reacties, hoe eenvoudiger het wordt om complex gedrag te benaderen, ten koste van extra systeemcomplexiteit.
Pad naar energiezuinig rekenen in het laboratorium
Voorbij de simulaties kijkt het artikel vooruit naar praktische apparaten. Het bespreekt hoe de keuze van reacties een evenwicht moet vinden tussen energieverbruik, bestuurbaarheid met enzymen of katalysatoren, en het vermogen om sleutelsoorten in realtime te meten, bijvoorbeeld met optische of elektrochemische methoden. De auteurs suggereren dat toekomstige microfluidische platforms zorgvuldig gekozen reactienetwerken zouden kunnen huisvesten, met ruimtelijke controle van inputs en ingebouwde detectie. Hoewel nog veel technische uitdagingen resteerden—van het omzetten van vergelijkingen naar echte chemie tot het omgaan met ruis en meetbeperkingen—laat de studie zien dat bescheiden reactiesystemen al de oplossingen van gekoppelde differentiaalvergelijkingen kunnen nabootsen. Voor de niet-specialist is de kernboodschap dat chemie zelf kan fungeren als een analoge computer, en daarmee een pad opent naar wetenschappelijke berekeningen die gebruikmaken van de energiezuinige processen die de natuur al miljarden jaren perfectioneert.
Bronvermelding: Johnson, C.G.M., Bohm Agostini, N., Cannon, W.R. et al. Energy-efficient scientific computing using chemical reservoirs. npj Unconv. Comput. 3, 17 (2026). https://doi.org/10.1038/s44335-026-00053-9
Trefwoorden: chemisch rekenen, energiezuinig rekenen, reservoir computing, chemische reactienetwerken, ordinary differential equations